









Serviços Personalizados
Journal

artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores
Compartilhar

 Permalink
PermalinkJournal of Human Growth and Development
versão impressa ISSN 0104-1282versão On-line ISSN 2175-3598
J. Hum. Growth Dev. vol.31 no.1 Marília jan./abr. 2021
https://doi.org/10.36311/jhgd.v31.11074
ORIGINAL ARTICLE
Brugada syndrome: current concepts and genetic background
Andrés Ricardo Pérez-RieraI; Joseane Elza Tonussi MendesI; Fabiola Ferreira da SilvaI; Frank YanowitzII; Luiz Carlos de AbreuI, VI, VII; José Luiz FigueiredoVIII; Rodrigo Daminello RaimundoI; Raimundo Barbosa-BarrosII; Kjell NikusIV; Pedro BrugadaV
ILaboratório de Delineamento de Estudos e Escrita Científica. Centro Universitário FMABC, Santo André, São Paulo, Brazil
IIIntermountain Medical Center, Intermountain Heart Institute, Salt Lake City, UT, United States; The University of Utah, Department of Internal Medicine, Salt Lake City, UT, United States
IIICoronary Center of the Hospital de Messejana Dr. Carlos Alberto Studart Gomes, Fortaleza, Ceará, Brazil
IVHeart Center, Tampere University Hospital and Faculty of Medicine and Health Technology, Tampere University, Finland
VScientific Director, Cardiovascular Division, Free University of Brussels (UZ Brussel) VUB, Brussels, Belgium
VIAdjunct Professor. School of Medicine. University of Limerick, Ireland
VIIProfessor. Department of Integrated Health Education and Graduate Program in Collective Health. Federal University of Espírito Santo, ES, Brazil
VIIIDepartment of Surgery, Experimental Surgery Unit, Federal University of Pernambuco, Recife, Pernambuco, Brazil
ABSTRACT
BACKGROUNG: Brugada syndrome (BrS) is a hereditary clinical-electrocardiographic arrhythmic entity with low worldwide prevalence. The syndrome is caused by changes in the structure and function of certain cardiac ion channels and reduced expression of Connexin 43 (Cx43) in the Right Ventricle (RV), predominantly in the Right Ventricular Outflow Tract (VSVD), causing electromechanical abnormalities. The diagnosis is based on the presence of spontaneous or medicated ST elevation, characterized by boost of the J point and the ST segment ≥2 mm, of superior convexity "hollow type" (subtype 1A) or descending rectilinear model (subtype 1B). BrS is associated with an increased risk of syncope, palpitations, chest pain, convulsions, difficulty in breathing (nocturnal agonal breathing) and/or Sudden Cardiac Death (SCD) secondary to PVT/VF, unexplained cardiac arrest or documented PVT/VF or Paroxysmal atrial fibrillation (AF) in the absence of apparent macroscopic or structural heart disease, electrolyte disturbance, use of certain medications or coronary heart disease and fever. In less than three decades since the discovery of Brugada syndrome, the concept of Mendelian heredity has come undone. The enormous variants and mutations found mean that we are still far from being able to concretely clarify a genotype-phenotype relationship. There is no doubt that the entity is oligogenetic, associated with environmental factors, and that there are variants of uncertain significance, especially the rare variants of the SCN5A mutation, with European or Japanese ancestors, as well as a spontaneous type 1 or induced pattern, thanks to gnomAD (coalition) researchers who seek to aggregate and harmonize exome and genome sequencing data from a variety of large-scale sequencing projects and make summary data available to the scientific community at large). Thus, we believe that this in-depth analytical study of the countless mutations attributed to BrS may constitute a real cornerstone that will help to better understand this intriguing syndrome.
Keywords: Brugada Syndrome, arrhythmic, environmental, genotype, phenotyp.
Authors summary
Why was this study done?
In less than three decades since the discovery of Brugada syndrome, the concept of Mendelian heredity has fallen apart. There is no doubt that the entity is oligogenetic associated with environmental factors.
What did the researchers do and find?
Recent research by the American College of Medical Genetics and Genomics / Association for Molecular Pathology (ACMG/AMP) has shown us that variants of uncertain significance, especially the rare variants of the SCN5A mutation, with European or Japanese ancestors, as well as spontaneous type 1 pattern or induced, thanks to Genome Aggregation Database (gnomAD) (coalition of researchers who seek to aggregate and harmonize exome and genome sequencing data from a variety of large-scale sequencing projects and make summary data available to the scientific community in generall).
What do these findings mean?
The enormous variants and mutations found mean that we are still far from being able to concretely clarify a genotype-phenotype relationship. Thus, we believe that this in-depth analytical study of the numerous mutations attributed to BrS can constitute a truly cornerstone that will help to better understand this intriguing syndrome.
INTRODUCTION
The Brugada Syndrome (BrS) is a hereditary clinical-electrocardiographic arrhythmic entity with a low prevalence worldwide (0.5 per 1,000 or 5 to 20 per 10,000 individuals), however, endemic in Southeast Asia (prevalence of 3.7 per 1,000). BrS clearly has male preponderance with a male/female ratio of 9:1 in Southeast Asia and 3:1 among Caucasians.
The syndrome is caused by alterations in the structure and function of certain cardiac ion channels and reduced expression of Connexin 43 (Cx43) in the Right Ventricle (RV), predominantly in the Right Ventricular Outflow Tract (RVOT) causing electromechanical abnormalities. The reduced and heterogeneous expression of Cx43 produces functionally significant electrophysiological heterogeneity in the ventricular wall and may promote transmural dispersion of repolarization. Until recently, BrS was considered an Autosomal Dominant (AD) Mendelian entity in ≈25% of cases or alternatively, sporadic.
It is currently thought that BrS most likely is an oligogenic disorder, rather than a Mendelian condition*, affecting several loci, and influenced by environmental factors. The diagnosis is based on the presence of a spontaneous or drug-induced ST elevation characterized by elevation of the J point and the ST segment of ≥2 mm, of superior convexity "coved type" (Subtype 1A) or descending rectilinear (Subtype 1B) type. The ST elevation is followed by a symmetric negative T wave in ≥1 right and/or high right precordial leads.
In the Subtype 1C, the J-point elevation is located to the inferior or inferolateral leads, with or without association with the early repolarization pattern. Brs is associated with an increased risk of syncope, palpitations, precordial pain, seizures, difficulty in breathing (nocturnal agonal respiration), and/or Sudden Cardiac Death (SCD) secondary to PVT/VF, unexplained cardiac arrest or documented PVT/VF or paroxysmal Atrial Fibrillation (AF) in the absence of macroscopic or apparent structural heart disease, electrolyte disturbance, use of certain drugs or coronary heart and fever.
The event typically occurs during the midnight-to-early-morning period at rest (≈80% of cases) or at a low level of physical activity especially during sleep, which suggests that parasympathetic tone is a determining factor in arrhythmogenesis: higher level of vagal tone and higher levels of Ito (cardiac transient outward potassium current) is evident during slower heart rates. Although BrS is considered a genetic disease, its mechanism remains unknown in ≈70-75% of cases and no single mutation is sufficient to cause the BrS phenotype. Although ≈20% of patients with BrS carry mutations in SCN5A, which encodes for the pore-forming α subunit of the cardiac sodium channels, the molecular mechanisms underlying this condition are still largely unknown. SCN5A, that was identified as the first BrS-associated gene in 1998, has emerged as the most common gene associated with the syndrome. The SCN5A gene is considered as the only gene definitely associated with BrS.
Currently, the oligogenic disease model is the accepted model1. More than 400 mutations in the SCN5A gene have been associated with SB. In an evidence-based review of genes reported to cause BS, which are in clinical use, 20 of the 21 genes did not have enough genetic evidence to support their causality for BS.
Type 2 Brugada ECG (Electrocardiographic/Electrocardiogram) pattern has also been associated with mutations in SCN5A (glycerol-3-phosphate dehydrogenase 1-like (GPD1L) protein), which is the domain responsible for a site homologous to SCN5A, and CACNA1C, the gene responsible for the α-subunit of cardiac L-type calcium channels.
To date, mutations of more than 20 genes, other than SCN5A, have been implicated in the pathogenesis of BrS. Multiple pathogenic variants of genes have been shown to alter the normal function of sodium ↓Loss- Of-Function (↓LOF), potassium Gain-Of-Function (↑GOF), and mutations in genes encoding for potassium channels have also been implicated.
Genes influencing Ito, include KCNE3, KCND3 and SEMA3A (semaphoring, an endogenous potassium channel inhibitor) while KCNJ8, HCN4, KCN5 and ABCC9 (encoding for SUR2A, the ATP-binding cassette transporter for the KATP channel) mutations affected the ATP-sensitive potassium channel (or KATP channel). KCNH2, which encodes for IKr was proposed by Wang et al2 to be associated with the BrS.
Dysfunction in the KCNAB2, which encodes the voltage-gated potassium channel β2-subunit, was associated with increased Ito activity and identified as a putative gene involved in BrS. Kvβ2 dysfunction can contribute to the Brugada ECG pattern3.
Classification of hereditary diseases
o Monogenic or Mendelian: to be transmitted to the offspring according to Mendel's laws. They can be: 1) AD, 2) Autosomal Recessive (AR), or 3) X-linked. Mendelian inheritance refers to the patterns of inheritance that are characteristic of organisms that reproduce sexually. It refers to the type of inheritance that can be easily understood as a consequence of a single gene.
o Multifactorial or polygenic: produced by mutations in several genes, generally of different chromosomes and the combination of multiple environmental factors (age, sex, obesity, tobacco or alcohol use, toxic environments or a limited childhood).
o Oligogenic*: there are a few genes that have more influence than the rest. In the case of BrS, this is the case for the SCN5A gene. The inheritance also depends on the expression of other mutations (epistasis: https://academic.oup.com/hmg/article/11/20/2463/616080). Despite their importance, mutations in the SCN5A gene are present in ≈20 to 30% of cases.
Ancestral differences also have impact on the classification of pathogenicity of variants identified from BrS patients4. The causality of BrS-associated genes is much disputed; many of these genes demand further research, but may be clinically valid. Although controversies still exist, more than two decades of extensive research in BrS has helped researchers to gain a better understanding of the overall spectrum of the condition, including its molecular pathophysiology, genetic background, and management.
Sanger sequencing was considered as the gold standard for DNA sequencing, applied for the mutation screening of BrS5. With newtechnologies, such as microarrays, whole-exome sequencing, and whole genome sequencing, it is possible to identify a variant at a single nucleotide resolution in relatively medium- to large-sized genomic regions. These technological genomic advancements enable the detection of genetic variations in patients, with high accuracy and reduced cost6. Therefore, it is probably only a matter of time before the puzzle of genetics in BrS is solved7,8.
Whole-exome sequencing
This is a genomic technique for sequencing all of the protein-coding regions of genes in a genome (known as the exome). It consists of two steps: the first step is to select only the subset of DNA that encodes proteins. These regions are known as exons - humans have about 180,000 exons, constituting about 1% of the human genome, or approximately 30 million base pairs.
The second step is to sequence the exonic DNA using any high-throughput DNA sequencing technology9. The goal of this approach is to identify genetic variants that alter protein sequences, and to do this at a much lower cost than whole-genome sequencing. Since gene variants can be responsible for both Mendelian and common polygenic diseases, whole exome sequencing has been applied both in academic research and clinical practice.
Exome sequencing is especially effective in the study of rare Mendelian diseases, because it is an efficient way to identify the genetic variants in all of an individual's genes. These diseases are most often caused by very rare genetic variants that are only present in a tiny number of individuals10. By contrast, techniques such as SNP arrays, can only detect shared genetic variants that are common to many individuals in the wider population11.
Furthermore, because severe disease-causing variants are much more likely (but by no means exclusively) to be in the protein coding sequence12, focusing on this 1% costs far less than whole-genome sequencing but still detects a high yield of relevant variants. The traditional way of genetic diagnostics, where clinical genetic tests were chosen based on the clinical presentation of the patient (i.e. focused on one gene or a small number known to be associated with a particular syndrome), or surveyed based only on certain types of variation (e.g. comparative genomic hybridization), provided definitive genetic diagnoses in fewer than half of all patients13.
Exome sequencing is now increasingly used to complement these other tests: both to find mutations in genes already known to cause disease as well as to identify novel genes by comparing exomes from patients with similar clinical features.
Whole genome sequencing
Whole genome sequencing is ostensibly the process of determining the complete DNA sequence of an organism's genome at a single time. This entails sequencing all of an organism's chromosomal DNA as well as DNA contained in the mitochondria and, for plants, in the chloroplast. In practice, genome sequences that are nearly complete are also called whole genome sequences.
Whole genome sequencing has largely been used as a research tool, but was introduced into the clinics in 201410,11,13.
In the future of personalized medicine, whole genome sequence data may be an important tool to guide therapeutic approach14. The tool of gene sequencing at single nucleotide polymorphism (SNP) level is also used to pinpoint functional variants from association studies and improve the knowledge available to researchers interested in evolutionary biology, and hence may lay the foundation for predicting disease susceptibility and drug response.
Whole genome sequencing should not be confused with DNA profiling, which only determines the likelihood that genetic material came from a particular individual or group, and does not contain additional information on genetic relationships, origin or susceptibility to specific diseases15. In addition, whole genome sequencing should not be confused with methods that sequence specific subsets of the genome - such methods include whole exome sequencing (1-2% of the genome) or SNP genotyping (<0.1% of the genome).
As of 2017, there were no complete genomes for any mammals, including humans. Between 4% to 9% of the human genome, mostly satellite DNA, had not been sequenced (https://www.statnews.com/2017/06/20/human-genome-not-fully-sequenced/).
Stringent variant interpretation guidelines can lead to high rates of Variants of Uncertain Significance (VUS) for genetically heterogeneous disease like LQTS and BrS. Quantitative and disease-specific customization of American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines can address this false negative rate.
The authors compared rare variant frequencies from 1847 LQTS (KCNQ1/KCNH2/SCN5A) and 3335 BrS (SCN5A) cases from the International LQTS/BrS Genetics Consortia to population-specific Genome Aggregation Database (gnomAD) data and developed disease-specific criteria for ACMG/AMP evidence classes-rarity (PM2/BS1 rules) and case enrichment of individual (PS4) and domain-specific (PM1) variants.
Rare SCN5A variant prevalence differed between European (20.8%) and Japanese (8.9%) BrS patients and diagnosis with spontaneous (28.7%) versus induced (15.8%) Brugada type 1 ECG (Electrocardiographic/Electrocardiogram). Ion channel transmembrane regions and specific N-terminus (KCNH2) and C-terminus (KCNQ1/KCNH2) domains were characterized by high enrichment of case variants and >95% probability of pathogenicity. Applying the customized rules, 17.4% of European BrS and 74.8% of European LQTS cases had (likely) pathogenic variants, compared with estimated diagnostic yields (case excess over gnomAD) of 19.2%/82.1%, reducing VUS prevalence to close to background rare variant frequency.
The authors concluded that large case-control data sets enable quantitative implementation of ACMG/AMP guidelines and increased sensitivity for inherited arrhythmia genetic testing16.
Brugada syndrome -susceptibility genes
Brs-1 brugada syndrome 1; brgda117
Locus: 3p21-23; OMIM: 601144; Gene: SCN5A. Only the SCN5A gene is classified as having definitive evidence as a cause for BrS18; Ion channel and effect: INa+↓LOF; Protein: Nav1.5- α subunit of the cardiac sodium channel carrying the sodium current INa+; Proportion of BrS attributed to this genetic variant: 11-28%. Phenotypes: Mutations in SCN5A lead to a broad spectrum of phenotypes, however the SCN5A gene is not commonly involved in the pathogenesis of BrS and associated disorders. Studies have revealed significant overlap between aberrant rhythm phenotypes, and single mutations have been identified that evoke multiple rhythm disorders with common gating lesions19. Figure 1 shows the numerous phenotypes with SCN5A mutations.
↑GOF mutations in SCN5A lead to more sodium influx into the cardiomyocytes through aberrant channel gating and cause Congenital long QT syndrome variant 3 (LQT3)20-22.
↓LOF mutations in SCN5A lead to lower levels of expression of SCN5A or production of defective Nav1.5 proteins, thereby causing BrS.
↓LOF and ↑GOF mutations may cause Dilated Cardiomyopathy (DCM).
Other SCN5A-related diseases are: Multifocal Ectopic Purkinje-related Premature Contractions (MEPPC) (↑GOF mutations)23, isolated cardiac conduction defect (↓LOF mutations)24, Sick Sinus Syndrome (SSS) (↓LOF mutations), Familial Atrial Fibrillation (FAF) (↓LOF or ↑GOF mutations), and overlap syndromes (both ↓LFO and ↑GOF mutations). Growing insights into the role of SCN5A in health and disease has enabled clinicians to lay out gene-specific risk stratification schemes and mutation-specific diagnostic and therapeutic strategies in the management of patients with a SCN5A mutation25.
Based on a study of AF in a large cohort of BrS patients, Amin et al.,26 hypothesized that a reduced number of potentially triggering Premature Atrial Contractions (PACs) in the presence of a more extensive substrate in SCN5A mutation carriers may explain the fact that AF is not more prevalent in patients with SCN5A mutations than in those without. Given the polemic and complex issues underlying the pathophysiology of BrS, one should regard this hypothesis as one potential mechanism of many that influence the prevalence of AF in BrS.
Figure 2 shows the sarcolemma cytoplasmic membrane crossed by the sodium channel, the components of this channel and the temporal correlation between the AP and the surface ECG (Electrocardiographic/Electrocardiogram).
Mutations in the SCN5A gene can produce ↑GOF, ↓LOF or both in the sodium channel (figure 3).
Overlap syndromes resulting from genetic defects caused by ↓LOF of sodium channel current (INa) or ↑GOF Late INa. In the absence of prominent Ito or IK-ATP, ↓LOF mutations in the inward currents result in various manifestations of PCCD. In the presence of prominent Ito or IK-ATP, ↓LOF mutations in inward currents cause conduction disease as well as J-wave syndromes (BrS and ERS). ↓LOF mutations of inward current in the presence of prominent ITO in certain regions of the Left Ventricle (LV), particularly the inferior wall, are considered etiologic factors of ERS (Early Repolarization Syndrome). The genetic defects that contribute to BrS and ERS can also contribute to the development of LQT3 and PCCD, in some cases causing multiple expressions of these overlap syndromes. In some cases, structural defects contribute to the phenotype, such as DCM, Left Ventricular Non compaction (LVNC).
More than 400 mutations have been identified in the SCN5A gene. Although the mechanisms of SCN5A mutations leading to a variety of channelopathies can be classified according to the alteration of INa-P and INa-L acting thorugh ↑GOF or ↓LOF, few researchers have summarized the mechanisms in this way27. ↑GOF mutations in SCN5A lead to more sodium influx into the cardiomyocytes through aberrant channel gating causing LQT3. Slowed or incomplete inactivation of the NaV1.5 channel results in an additional inward current, known as the late or persistent sodium current (Ipst), during the plateau phase of the ventricular action potential with ST segment prolongation and late T occurrence. Among the mutations in SCN5A associated with LQT3 is 1795insD, which is characterized by the insertion of 3 nucleotides (TGA) at position 5537 C-terminal domain of the NaV1.5 protein20. Carriers of this mutation may not only present with LQT3, but also with ECG (Electrocardiographic/Electrocardiogram) features of sinus bradycardia, PCCD, and BrS, thus creating the first described arrhythmic 'overlap syndrome28.
SCN5A 1795insD is supposed to be a ↑GOF mutation in light of the QT prolongation. A ↓LOF mutation cause sinus bradycardia, PCCD, and BrS. MEPPC is caused by both ↓LOF and ↑GOF mutations. ↓LOF in SCN5A result in amplitude reduction in the peak sodium current, leading to channel protein dysfunction or PCCD an entity with minor structural heart disease. Both↓LOF and ↑GOF mutations may cause DCM and/or AF25. ↑GOF MEPPC is a rare cardiac syndrome combining Polymorphic Ventricular Tachycardia (PVT) with DCM23.
Experimental animal models have shown that there is a potential role for Purkinje fibers in the initiation of arrhythmias in different disease entities, such as LQT3 (SCN5A), Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) (RyR2), and Idiopathic Ventricular Fibrillation (IVF) (DPP6). This is also the case in experimental clinical scenarios in MEPPC (SCN5A), IVF (DPP6)29. Recently, Haïssaguerre et al.,30 found two potential explanations in arrhythmic SCD without apparent structural heart disease: a microstructural background (fibrosis, inflammation, fat infiltration, dysplasia, etc) or truly electrical heart disease in the myocardium or the Purkinje fibers. They highlighted three possible mechanisms: 1) focal excitation abnormality (IVF, CPVT, MEPPC); 2) dromotropic disturbance (BrS, J wave syndrome, IVF); and 3) repolarization abnormality (Long QT syndrome [LQTS], short QT syndrome [SQTS], ERS)30.
In the ECG (Electrocardiographic/Electrocardiogram), PR interval prolongation is the only parameter that predicted the presence of a SCN5A mutation in BrS.
Late potentials in high resolution ECG were more frequently observed in SCN5A mutation carriers31.
SCN5A mutation is associated with an increased risk of drug-induced ventricular arrhythmia in patients without baseline type-1 BrS ECG. In particular, Snon-missense and Smissense-TP represent high risk32.
The genetic basis of BrS is poorly understood, and there is growing evidence that many cases are polygenic, with familial inheritance rarely reported. Genetic testing in the clinical setting should be carefully considered and confined to SCN5A. Patients with a positive family history of disease, a spontaneous type 1 ECG pattern and symptomatic presentations may have a greater genetic yield though this is an area requiring further research.
Polygenic diseases are caused by numerous genetic and non-genetic factors, with coronary artery disease being a well-described example. By performing genome-wide association studies (GWAS), thousands of single nucleotide variants which each provide a small incremental increased risk of disease can be identified33. A polygenic risk score (PRS) is the predicted additive effect of these variants which can be used to predict the risk of developing disease. While not currently in clinical practice, the study reported by Khera and colleagues illustrated the potential for PRS to provide important prognostic value in the management of patients with coronary artery disease34. By combining millions of common variants, a PRS was calculated showing better prediction of coronary artery disease than any single traditional risk factor. While promising, these findings are yet to be associated with clear clinical interventions, and to date have the greatest predictive power in European individuals. In the future, used in tandem with clinical investigations and information, PRS may enhance our ability to predict those at greater risk of complex diseases or adverse outcomes, even further necessitating access to specifically trained genetic counsellors.
Mutations in SCN5A, GPDIL, KCND3 and KCNJ8, Kir6.1 genes s cause ↓LOF on Na+↓INa+/and ↓Ca++ and ↑GOF in ↑ITO ↑IK=ATP channels affecting epi-/endomyocardial APs, resulting in the type 1 BrS ECG) pattern, triggers of short coupled PVCs and eventually VT/VF (figure 5).
Micaglui et al., presented a case report, with a novel inherited frameshift mutation (c.4700_4701del (p. Phe1567Cysfs*221) in a single copy of the SCN5A gene associated with BrS. The proband experienced ventricular events successfully treated with DC-shock and he also suffered from supraventricular tachycardia. An ajmaline test confirmed the BrS diagnosis. The same mutation was found in the father and sister, who were both diagnosed with BrS. The authors hypothesize that this SCN5A frameshift mutation could be responsible for BrS and potentially linked to supraventricular tachycardias35.
Monaski et al., reported the novel NM_198056.2:c.1111C>T (p.Gln371*) heterozygous variant in the SCN5A gene, as well as its segregation with BrS in a large family. The proband was an Italian young adult woman, who had previously performed genetic counseling elsewhere with a positive test for the familial heterozygous variant in the SCN5A gene. She complained of palpitations. She underwent an ajmaline challenge, which was positive. Later on, she had PVT, underwent an Electrophysiological Study (EPS), which was positive, and received an Implantable Cardioverter Defibrillator (ICD). The proband then underwent RF of the AS, which was found in the epicardium of the RVOT. Ajmaline was administered prior to ablation, suggesting a pathogenic effect of this variant36.
Monaski et al., reported the largest family to-date with the segregation of the heterozygous variant NM 198056:c.4894C>T (p.Arg1632Cys) in the SCN5A gene. The genotype-phenotype relationship observed suggests a likely pathogenic effect of this variant. Functional studies to better understand the molecular effects of this variant are necessary37.
Micaglio et al., reported for the first time a family in which the inherited nonsense mutation [c. 3946C > T (p.Arg1316*)] in the SCN5A gene segregated in association with BrS. Moreover, they also reported, for the first time, the frameshift mutation [c.7686delG (p.Ile2563fsX40)] in the NF1 gene, as well as its association with type 1 neurofibromatosis (NF1). Both of these mutations and associated phenotypes were discovered in the same family. This genetic association may identify a subset of patients at higher risk of SCD. This case series highlights the importance of genetic testing not only to confirm the molecular pathology but also to identify asymptomatic family members, who need clinical examinations and preventive interventions, as well as to advise about the possibility of avoiding recurrence risk with medically assisted reproduction38.
Micaglio et al., presented a 30-year-old Italian male proband with a history of palpitations and syncope since puberty. He received a diagnosis of BrS elsewhere due to a spontaneous type 1 BrS ECG pattern. His brother had also been diagnosed with BrS, and had an ICD implanted. Prior to radiofrequency ablation of the arrhythmogenic substrate, genetic testing revealed the variant NM_198056.2:c.2091G>A (p.Trp697X) in the SCN5A gene (Leiden Open Variation. A spontaneous type 1 ECG was observed. The novel heterozygous variant NM_198056.2:c.2091G>A (p.Trp697X) in the SCN5A gene segregates with BrS in the family presented, providing crucial human data relevant to understanding the pathology of BrS for patients with this variant. The study results suggested a likely pathogenic effect of the variant and could be used as a stepping stone for functional studies to better understand the molecular pathways involved39.
Smani et al., reported the SCN5A mutation L1393X, identified in a patient with overlap phenotypes PCCD and BrS and SCN5A‐E1784K and SCN5A‐H558R polymorphism causing Overlapping Phenotype of Long QT Syndrome, BrS, and Conduction Defect40.
Eight SCN5A nonsense mutations were identified in BrS patients, L1393X41 did not form functional channels, which may severely affect the electrophysiological properties of the heart. Interestingly, none of those eight nonsense mutations have been reported to cause PCCDs. Also, the proband patient did not show severe BrS phenotypes (i.e., no syncope or aborted SCD) despite the severe functional defects of sodium channels. However, this might be due to the timing of diagnosis. Probst reported that conduction defects can be developed over time in the patients who carry the SCN5A mutations42. In addition, some patients with the spontaneous type 1 Brugada ECG pattern at rest or during drug-challenge tests experience severe ventricular events, whereas other patients or family members carrying the same mutations sometimes remain asymptomatic43. Since this is a single case report, there is a limitation in linking this experimental data to the clinical scenario41.
There are four known common polymorphisms of the SCN5A gene related to BrS, including R34C, H558R, S1103Y, and R1193Q44,45. These polymorphisms could decrease expression of sodium‐channel proteins and alter gating properties resulting in prolonged QRS duration and slow conduction in the heart46. The SCN5A mutations may be associated with early and frequent Ventricular Fibrillation (VF) recurrence or Sudden Cardiac Arrest (SCA) in BrS patients, which may be related to fibrosis in the epicardial surface of the right ventricular outflow tract47. SCN5A‐R1193Q is a genetic marker associated with cardiac conduction defects and VF in symptomatic BrS patients.
Table 2 shows inherited cardiomyopathies and inherited arrhythmia syndromes with robust evidence for disease causation and figure 1 assesses the weight of evidence for pathogenicity using Gnomad: Genome Aggregation Database.
BrS-2 BRUGADA SYNDROME 2; BRGDA2
Locus: 12p13.3; OMIM: #611777; Gene: GPD1L; Ion channel and effect: ↓INa+ LOF; Protein: Glycerol-3phosphate dehydrogenase 1-like protein. Peptide-reduced GPD1-L activity leads to phosphorylation of Nav1.5 and decreased ↓INa+ Channels affected and effect: ↓INa+ LOF. Amino acid substitution A280V49; Proportion of BrS attributed to this variant: Rare <1%50. Other Phenotypes: Defects in this gene may also cause Sudden Infant Death Syndrome sometimes known as "cot death" (SIDS), sometimes known as "cot death" - sudden, unexpected and unexplained death of an apparently healthy infant less than one year old despite thorough case investigation, including complete autopsy, examination of the death scene, and review of clinical history51.
BrS-3 BRUGADA SYNDROME 3; BRGDA3
Locus: 12p13.33, which is the short (p) arm of chromosome 12 at position 13.33; OMIM: 114205; Gene: CACNA1C, Cav1.2; Ion channel and effect: ↓Ica2+ LOF; Protein: Voltage-dependent L-type calcium channel subunit α-1C/Cav1.2- a-subunit of the voltage-gated calcium channel carrying the L-type Ca2+ current ICa(L); Proportion of BrS attributed to this genetic variants: 6.6%; Amino acid Substitution: A39V, G490R; Other phenotypes: Timothy syndrome (TS1), SN5A and CACNA1C, complex BrS52, congenital cardiac anomalies, cardiomyopathy, neonatal onset epileptic encephalopathy53, bipolar type I disorder54, schizophrenia55.
BrS-4 BRUGADA SYNDROME 4; BRGDA4
Locus: 10p12.33; OMIM: 600003; Gene: CACNB2b, Cavβ2b; Ion channel and effect: ↓ICa++ LOF; Protein: Voltage-dependent L-type calcium channel subunit β-2 or Cavβ2B- β-2 subunit of the voltage-gated calcium channel carrying the L-type calcium current ICaL (LTCC) regulates calcium entry into cardiomyocytes. CACNB2 (β2) LTCC auxiliary subunits traffic the pore-forming CACNA subunit to the membrane and modulate channel kinetics. β2 is a membrane associated guanylate kinase (MAGUK) protein; Proportion of BrS attributed to this genetic variants: 4.8%.
Note: A major role of MAGUK proteins is to scaffold cellular junctions and multiprotein complexes. β2.1 may also function in the heart as a MAGUK scaffolding unit to maintain N-cadherin-based adherens junctions and heart tube integrity56 protein is required in the heart for control of cell proliferation and heart tube integrity56.
BrS-5 BRUGADA SYNDROME 5; BRGDA5
Locus: 19q13,1; OMIM: 600235; Gene: SCN1B, Na↓1; Ion channel and effect: ↓INa+ ↓LOF. Heterologous expression assays illustrate that reported SCN1B mutations are ↓LOF and appear to interfere with modulation of channel gating57; Protein: Navβ1. Sodium channel subunit β-1 is a protein that in humans is encoded by the SCN1B gene: INa+.
This is proportion of BrS attributed to this genetic variants: 4.8%: 1.1%; Phenotypes: Mutation in the SCN1B gene are associated with BrS and generalized epilepsy with febrile seizures plus (GEFS+). GEFS+ is a syndromic autosomal dominant disorder where afflicted individuals can exhibit numerous epilepsy phenotypes. GEFS+ can persist beyond early childhood (i.e., 6 years of age). GEFS+ is also now believed to encompass three other epilepsy disorders: severe myoclonic epilepsy of infancy (SMEI) or Dravet's syndrome, borderline SMEI (SMEB), and intractable epilepsy of childhood (IEC)58. ↓LOF mutations in the β-subunits (encoded by C) have also been described for AF59.
OMIN: # 612838. A number sign (#) is used with this entry because of evidence that BrS-5 and a nonspecific cardiac conduction defect are caused by heterozygous mutation in the SCN1B gene on chromosome 19q13,1. Other non-cardiac entities related to SCN1B mutations: The mutation C121W occurred in temporal lobe epilepsy60, and a single SCN1B mutation has been reported in Dravet syndrome (a rare, catastrophic, lifelong form of epilepsy that begins in the first year of life with frequent and/or prolonged seizures)61.
BrS-6 BRUGADA SYNDROME 6; BRGDA6
Locus: 11q13-14; OMIM: 604433; Gene: KCNE3, MiRP2; Ion channel and affect: ↑Ito in phase 1 of the action potential. ↑GOF; Protein: Potassium voltage -gated channel subfamily E member 3 MiRP2- β subunit to voltage potassium channels. Modulates the transient outward potassium current Ito; Proportion of BrS attributed to this genetic variants: <1% (rare). # 613119 A number sign (#) is used with this entry because of evidence that BrS-6 is caused by heterozygous mutation in the KCNE3 gene on chromosome 11q13.
Delpont et al., studied 105 probands with the BrS, who were screened for ion channel gene mutations using single-strand conformation polymorphism electrophoresis and direct sequencing. A missense mutation (R99H) in KCNE3 (MiRP2) was detected in one proband. The R99H mutation was found 4/4 phenotype-positive and 0/3 phenotype-negative family members. Chinese hamster ovary-K1 cells were cotransfected using wild-type (WT) or mutant KCNE3 and either WT KCND3 or KCNQ1. Whole-cell patch clamp studies were performed after 48 hours. Interactions between Kv4.3 and KCNE3 were analyzed in coimmunoprecipitation experiments in human atrial samples.
Cotransfection of R99H-KCNE3 with KCNQ1 produced no alteration in tail current magnitude or kinetics. However, cotransfection of R99H KCNE3 with KCND3 resulted in a significant increase in the Ito intensity (↑GOF Ito) compared with WT KCNE3KCND3.
Using tissues isolated from the left atrial appendages of human hearts, the authors also demonstrated that Kv4.3 and KCNE3 can be coimmunoprecipitated. These results provide evidence for a functional role of KCNE3 in the modulation of Ito in the human heart and suggest that mutations in KCNE3 can underlie the development of the BrS62. A KCNE3 T4A mutation was identified in a Japanese patient presenting with the Brugada ECG-pattern and neurally mediated syncope (NMS). Its functional consequence was the ↑GOF of Ito, which could underlie the pathogenesis of this ECG pattern. The data provide novel insights into the genetic basis of Japanese BrS63.
BrS-7 BRUGADA SYNDROME 7; BRGDA7
Locus: 11q23.3; OMIM: 6081214; Gene: SCN3B; Ion channel and affected: ↓INa ↓LOF64,65; Protein: Ran guanine nucleotide release factor; Proportion of BrS attributed to this genetic variants: <1% (rare).
Note: Navb-3 subunit of the cardiac sodium channel carrying the sodium current INa+; # 613120 A number sign (#) is used with this entry because of evidence that BrS-7and AF-16 (18, 19) are caused by heterozygous mutation in the SCN3B gene on chromosome 11q24.
The Val110Ile mutation of SCN3B is a relatively common cause of SCN5A-negative BrS in Japan, and has a reduced ↓ INa current because of the loss of cell surface expression of Nav1.566.
BrS-8 BRUGADA SYNDROME 8; BRGDA8
Locus: 12q11. 23; OMIM: 600935, Gene: KCNJ8; Protein: Kir6.1; Ion channel and effect: ↑IK=ATP ↑GOF; Protein: ATP-sensitive inward rectifier potassium channel 8 Kir6, carries the inward rectifier potassium current.
This is proportion of BrS attributed to this genetic variants: 2%. # 613123. A number sign (#) is used with this entry because of evidence that BrS-8 is caused by heterozygous mutation in the HCN4 gene on chromosome 15q2467; Authors: KCNJ8 is a susceptibility gene for BrS and ERS (Early Repolarization Syndrome) and point to S422L as a possible hotspot mutation. The S422Linduced ↑GOF in ↑IK=ATP sensitive potassium channel current is due to reduced sensitivity to intracellular ATP68.
BrS-9 BRUGADA SYNDROME 9; BRGDA9
Locus: 7q21.11; OMIM: 114204; Gene: CACNA2D1, Cavα2δ; Ion channel and effect: ↓ICa++ LOF; Protein: Voltage-dependent calcium channel subunit α2/δ1 subunit of the voltage-gated calcium channel carrying the L-type calcium current ICaL; Proportion of BrS attributed to this genetic variants: 1.8%. # 616399 A number sign (#) is used with this entry because of evidence that BrS-9 is caused by heterozygous mutation in the KCND3 gene on chromosome 1p1369.
Other phenotypes: Short QT syndrome type 6 (SQT6) was identified in a 17-year-old female who suddenly lost consciousness. VF was terminated by defibrillation. In hospital, her ECG showed a short QTc interval (329 ms) and tall, narrow T waves70. Programmed electrical stimulation could elicit AF and Ventricular Tachycardia (VT). Genetic screening revealed a S755T substitution CACNA2D1 encoded Cavα2δ-1 subunit of the L-type Ca2+ channel. Coexpression of the mutant Cavα2δ-1 subunit with Cav1.2α1 and Cavβ2b led to reduced ICa,L (using Ba2+ ions as charge carrier) compared to the WT control, without an obvious effect on surface expression, suggestive of a modification of single channel properties by the S755T Cavα2δ-171 and malignant hyperthermia susceptibility72.
BrS-10 BRUGADA SYNDROME 10; BRGDA10
Locus:1p13.2; OMIM: 605411; Gene: KCND3, Kv4.3; Ion channel and effect: ↑Ito ↑GOF for the phase-1 repolarization of the action potential; Protein: Potassium voltage-gated channel subfamily D member 3. Kv4.3, α-subunit of the transient outward potassium channel Ito. An increased Ito may directly affect cardiac conduction. However, the effects of an increased Ito on AP upstroke velocity or sodium current at the cellular level remain unknown.
This is proportion of BrS attributed to this genetic variants: <1%. There is a prominent role of the Ito in BrS pathogenesis, the rare ↑GOF mutations in KCND3 serve as a pathogenic substrate for BrS. Giudicessi et al, provided the first molecular and functional evidence implicating novel KCND3 ↑GOF mutations in the pathogenesis and phenotypic expression of BrS, with the potential for a lethal arrhythmia being precipitated by a genetically enhanced I(to) current gradient within the right ventricle where KCND3 expression is highest69.
Portero et al.,73 investigated the consequences of KV4.3 overexpression on NaV1.5 current and consequent sodium channel availability. They found that overexpression of KV4.3 protein in HEK293 cells stably expressing NaV1.5 (HEK293-NaV1.5 cells) significantly reduced NaV1.5 current density without affecting its kinetic properties. In addition, KV4.3 overexpression decreased AP upstroke velocity in HEK293-NaV1.5 cells, as measured with the alternating voltage/current clamp technique.
These effects of KV4.3 could not be explained by alterations in total NaV1.5 protein expression. Using computer simulations employing a multicellular in silico model, the authors demonstrated that the experimentally observed increase in KV4.3 current and concurrent decrease in NaV1.5 currents may result in a dromotropic disturbance, underlining the potential functional relevance. This study gives the first proof of concept that KV4.3 directly impacts on NaV1.5 currents73.
Giudicessi et al.,69. provided the first molecular and functional evidence implicating novel KCND3 ↑GOF mutations in the pathogenesis and phenotypic expression of BrS, with the potential for a lethal arrhythmia being precipitated by a genetically ↑Ito current gradient within the RV where KCND3 expression is the highest69.
BrS-11 BRUGADA SYNDROME 11; BRGDA11
Locus: 17p13.1; OMIM: 607954; Gene: RANGRF; Ion channel and effect: ↓INa+ ↓LOF; Protein: Sodium channel subunit beta 3that encodes MOG1 - influences trafficking of Nav 1.5. The protein MOG1 is a cofactor of the cardiac sodium channel, Nav1.5. Overexpression of MOG1 in Nav1.5-expressing cells increases sodium current markedly. Mutations in the genes encoding Nav1.5 and its accessory proteins have been associated with cardiac arrhythmias of significant clinical impact
This is proportion of BrS attributed to this genetic variants: <1% (rare)74; Olesen et al., while screening for the Nav1.5 cofactor MOG1, uncovered a novel nonsense variant, that appeared to be present more frequently among patients than in control subjects75.
In cardiomyocytes, MOG1 is mostly localized in the cell membrane and co-localized with Nav1.5. MOG1 is a critical regulator of cardiac sodium channel function. Wu et al, demonstrated the functional diversity of Nav1.5-binding proteins, which have important functions for Nav1.5 under different cellular conditions76.
Chakrabarti et al.,75,77, screening for the Nav1.5 cofactor MOG1, uncovered a novel nonsense variant that appeared to be more frequent among patients than control subjects. This variant causes MOG1 ↓LOF and therefore, it might be disease causing or modifying under certain conditions75,77.
BrS-12 BRUGADA SYNDROME 12; BRGDA12
Locus: 3p21.2-2-p14.3; OMIM: 602701; Gene: SLMAP; Ion channel and effect: ↓INa+ ↓LOF; Protein: Sarcolemma membrane-associated protein (SLMAP), a component of T-tubes and the sarcoplasmic reticulum - influences trafficking of Nav1.5;
The proportion of BrS attributed to this genetic variants: Rare. T-tubules and sarcoplasmic reticulum are essential in excitation of cardiomyocytes, and SLMAP is a protein of unknown function localizing at T-tubules and sarcoplasmic reticulum. This protein belongs to the super family of tail anchored membrane proteins which serve diverse roles including cell growth, protein trafficking and ion channel regulation. Three main SLMAP isoforms (SLMAP1 (35 kDa), SLMAP2 (45 kDa), and SLMAP3 (91 kDa)) are expressed in the myocardium but their precise role is unknown78.
The mutations in SLMAP may cause BrS via modulating the intracellular trafficking of hNav1.5 channel79.
BrS-13 BRUGADA SYNDROME 13; BRGDA13
Locus:12p12.1; OMIM: 601439; Gene: ABCC9 SUR2A; Ion channel and effect: ↑IK(ATP) ↑GOF; Protein: SUR2A, the adenosine triphosphate (ATP) binding cassette transporter of the IK(ATP) channel; Proportion of BrS attributed to this genetic variants: Rare. The ABCC9 is an ion channels/ion channel-related AF.
Adenosine triphosphate (ATP)-sensitive potassium cardiac channels consist of inward-rectifying channel subunits Kir6.1 or Kir6.2 (encoded by KCNJ8 or KCNJ11) and the sulfonylurea receptor subunits SUR2A (encoded by ABCC9). KCNJ8 is a susceptibility gene for BrS and ERS (Early Repolarization Syndrome) and point to S422L as a possible hotspot mutation.
The S422L-induced ↑GOF in ATP-sensitive potassium channel current is due to reduced sensitivity to intracellular ATP. ABCC9 has ERS and BrS susceptibility genes. A ↑GOF in IK-ATP when coupled with a ↓LOF in SCN5A may underlie type 3 ERS, which is associated with a severe arrhythmic phenotype68,80.
BrS-14 BRUGADA SYNDROME 14; BRGDA14
Locus: 11q23; OMIM: 601327; Gene: SCN2B, Navβ2; Ion channel and effect: ↓INa+ LOF; Protein: Sodium channel subunit β-2. Navβ2β -2subunit of the cardiac sodium channel carrying the sodium current ↓INa+; Proportion of BrS attributed to this genetic variants: <1% (rare).
Riuró et al.,59 identified a novel missense mutation in the sodium β2 subunit encoded by SCN2B, in a woman diagnosed with BrS. They studied the sodium current from cells coexpressing Nav 1.5 and wild type (β2WT) or mutant (β2D211G) β2 subunits. Electrophysiological analysis showed a reduction in INa+ density when Nav 1.5 was coexpressed with β2D211G.
Single channel analysis showed that the mutation did not affect the Nav 1.5 unitary channel conductance. Instead, protein membrane detection experiments suggested that β2D211G decreases Nav 1.5 cell surface expression.
The effect of the mutant β2 subunit on the INa strongly suggests that SCN2B is a candidate gene associated with BrS81.
Other phenotypes: FAF59. Genetic deletion of SCN2B in mice resulted in ventricular (VAs) and atrial arrhythmias, consistent with reported SCN2B mutations in human patients82. ↓INa+ LOF mutations were identified in patients with FAF and were associated with a distinctive ECG phenotype. Decreased Na+ current enhances AF susceptibility.
BRUGADA15 BRUGADA SYNDROME 15; BRGDA15
BrS15: Locus: 12p11; OMIM: 602861; Gene: PKP2; Protein: Desmosome protein Plakophillin-2; Ion channel and effect: ↓INa+ LOF; Proportion of BrS attributed to this genetic variants: <1% (rare).
Desmosome protein Plakophillin-2 interacts with INa+. Plakophilin-2 (PKP2) variants could produce a BrS phenotype, which is the same allelic disorder as some Sudden Unexplained Nocturnal Death Syndromes (SUNDS) variants.
All coding regions of the PKP2 gene in 119 SUNDS victims were genetically screened using PCR and direct Sanger sequencing methods. Three novel mutations (p.Ala159Thr, p.Val200Val, and p.Gly265Glu), one novel rare polymorphism (p.Thr723Thr), and eight polymorphisms were identified.
A compound mutation (p.Ala159Thr and p.Gly265Glu) and a rare polymorphism (p.Thr723Thr) were found in one SUNDS case with absence of the apparent structural heart disease. The detected compound mutation identified in this first investigation of the PKP2 genetic phenotype in SUNDS was regarded as the plausible genetic cause of this SUNDS case.
The rare incidence of PKP2 mutation in SUNDS (1%) supports the previous viewpoint that SUNDS is most likely an allelic disorder as BrS83. Mutations in proteins of the desmosome are associated with Arrhythmogenic Cardiomyopathy (AV). Life-threatening VAs often occur in the concealed forms/phases of the AC before the onset of structural changes.
It was suggested that loss of desmosomal integrity (including mutations or loss of expression of plakophilin-2; PKP2) leads to reduced sodium current, the PKP2-INa relation could be partly consequent to the fact that PKP2 facilitates proper trafficking of proteins to the intercalated disc, and, PKP2 mutations can be present in XV patients diagnosed with BrS thus supporting the previously proposed notion that AC and BrS are not two completely separate entities84.
Mutations on PKP2 account for the majority of AC cases, a disease characterized by high incidence of VAs and a progressive cardiomyopathy with fibrofatty infiltration involving predominantly the RV. Although BrS was initially described as a purely electric condition in intact hearts, it is now recognized that structural changes occur mainly at the RVOT85.
These findings support the hypothesis, suggested in the past by some clinicians, that the two conditions could be at the bookends of a phenotypical common spectrum. PKP2 is a structural protein of the desmosome whose principal role is to maintain tissue integrity and cell-to-cell stability.
However, data from cellular and mouse models demonstrated that loss of PKP2 could facilitate arrhythmias by decreasing sodium current86 through an electrophysiological effect. Indeed, in vitro characterization of the PKP2 mutations detected in patients with a BrS phenotype showed a decreased sodium current, consistent with the clinical phenotype.
Super-resolution microscopy data showed that loss of PKP2 could affect proper trafficking of the sodium channel at the membrane, thus supporting the concept that proteins could have accessory roles aside from the primary one ascribed to them.
The role of the cardiac intercalated disc as a functional unit with both structural and electric regulatory functions has been opening new paths of investigations on the possible arrhythmogenic substrate in BrS47.
BrS-16 BRUGADA SYNDROME 16; BRGDA16
Locus: 3q28-q29; OMIM: 601513; Gene: FGF12 (Fibroblast Growth Factor); Ion channel and effect: ↓INa+ LOF; Protein: FHAF1 Fibroblast growth factor homologues factor-1- mutation decreases ↓INa+; Proportion of BrS attributed to this genetic variants: <1% (rare)87. Multilevel investigations strongly suggest that Q7R-FGF12 is a disease-associated BrS mutation. FHF effects on↓INa+and ↓ICa++channels are separable.
Most significantly, a study by Hennessey et established a new method to analyze effects of human arrhythmogenic mutations on cardiac ionic currents. On the basis of the recent demonstration that FGF homologous factors (FHFs; FGF11-FGF14) regulate cardiac ↓INa+ and ↓ICa++channel currents, FHFs are candidate BrS loci88. Mutation FGF12 also causes neonatal-onset epilepsy89.
BrS-17 BRUGADA SYNDROME 17; BRGDA17
Locus: 3p22.22; OMIM: 604427; Gene: SCN10A, Nav1.8; Ion channel and effect: ↓INa+ LOFβ; Protein: Nav1.8-α subunit of the neural sodium channel; Proportion of BrS attributed to this genetic variants: 5-16.7%.
Hu et al.,90 identified SCN10A as a major susceptibility gene for BrS, thus greatly enhancing our ability to genotype and risk stratify probands and family members. The SCN10A SNP V1073 is strongly associated with BrS90, and is expressed in the myocardium and the conduction system, suggesting a possible role in the electrical function of the heart91.
Mutations in the SCN10A gene cause ↓LOF in INa: Co-expression of SCN5A-WT with SCN10A-mutant cause a major ↓LOF in INa, in BrS patients90. It is necessary to conduct studies in larger populations to better understand the role of SCN10A in BrSand other genetic cardiac arrhythmias92.
Rare variants in the screened QRS-associated genes (including SCN10A) are not responsible for a significant proportion of SCN5A mutation negative BrS. The common SNP SCN10A V1073 was strongly associated with BrS and demonstrated loss of NaV1.8 function, as did rare variants in isolated patients93.
The expression of sodium channel Nav1.8 in cardiac nervous systems has been identified, and variants of SCN10A that encodes Nav1.8 contribute to the development of BrS by modifying the function of Nav1.5 or directly reducing the sodium current.
Fukuyama et al94., screened for the SCN10A gene using a high-resolution melting method and direct sequencing and compared the clinical characteristics among the probands with gene mutations in SCN10A, 6 probands with CACNA1C and 17 probands with SCN5A. They identified six SCN10A variant carriers (2.5%): W189R, R844H (in two unrelated probands), N1328K, R1380Q, and R1863Q.
Five were male. Four were symptomatic: one died following SCD age 35, one suffered VF, and two had recurrent syncope. Compared with BrS patients carrying SCN5A or CACNA1C mutations, although there were no significant differences among them, symptomatic patients in the SCN10A group tended to be older than those in the other gene groups94.
El-Battrawy et al used a cellular model of BrS to study SCN10A mutations using human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs). They observed that patient-specific hiPSC-CMs are able to recapitulate single-cell phenotype features of with SCN10A mutations and may provide novel opportunities to further elucidate the cellular disease mechanism95.
Gray et al94., observed that the lack of genotype-phenotype concordance among families, combined with the high frequency of previously reported mutations in the Genome Aggregation Database browser, suggests that SCN1B is not a monogenic cause of BrS or SADS96.
SCN10A mutation phenotypes: possible association between variants in the SCN5A (600163), SCN10A, and HEY2 (604674) genes and BrS, see 601144. Associations Pending Confirmation; BrS+ERS3, SCN10A is an important susceptibility gene for BrS and for other cardiac syndromes, including cardiac conduction defect, ERS (Early Repolarization Syndrome), AF, VT/VF, RBBB, and bradycardia. SCN10A is known to be involved in nociception97.
BrS-18 BRUGADA SYNDROME 18; BRGDA18
Locus: 6q22.3; OMIM: 604674; Gene: helix-loop-helix transcription factor HEY2 (transcriptional factor). Hey2 is specifically expressed in the embryonic mouse ventricles and is indispensable for ventricular myocyte differentiation, compartment identity and morphogenesis of the heart98.
However, how Hey2 transcription is precisely regulated in the heart remains unclear; Ion channel and effect: ↓INa+ LOF; Protein: Transcription factor identified in GWAS; Proportion of BrS attributed to this genetic variants: Rare.
Common variants of SCN5A, SCN10A, and HEY2 are associated with BrS99.
The association signals at SCN5A-SCN10A demonstrate that genetic polymorphisms modulating cardiac conduction can also influence susceptibility to cardiac arrhythmia. The implication of association with HEY2, supported by new evidence that this gene regulates cardiac electrical activity, shows that BrS may originate from altered transcriptional programming during cardiac development99.
The study by Veerman et al., uncovered a role of HEY2 in the normal transmural electrophysiological gradient in the ventricle and provided compelling evidence that genetic variation at 6q22.31 (rs9388451) is associated with BrS through a HEY2-dependent alteration of ion channel expression across the cardiac ventricular wall100.
Andreasen et al.,101 investigated whether three single-nucleotide polymorphisms (SNPs) (rs11708996; G>C located intronic to SCN5A, rs10428132; T>G located in SCN10A, and rs9388451; T>C located downstream to HEY2) at loci associated with BrS in a GWAS also were associated with AF. They concluded that the prevalence of three risk alleles previously associated with BrS was lower in AF patients than in patients free of AF, suggesting a protective role of these loci in developing AF101.
Nakano et al.,102 investigated relationships between genotypes of 3 single-nucleotide polymorphisms reported in a recent GWAS and BrS phenotypes. Their findings suggest that the HEY2 CC genotype may be a favorable prognostic marker for BrS, protectively acting to prevent VF presumably by regulating the repolarization current102.
BrS-19 BRUGADA SYNDROME 19; BRGDA19
Locus: 7p12.1; OMIM: 603961; Gene: SEMA3A, semaphoring; Ion channel and effect: Ito ↑GOF; Protein: NaV1.5 - α subunit of the cardiac sodium channel carrying the sodium current INa; Proportion of BrS attributed to this genetic variants: <1% (rare). Boczek et al.103 were the first to demonstrate SEMA3A as a naturally occurring protein that selectively inhibits Kv4.3 and SEMA3A as a possible BrS susceptibility gene through a Kv4.3 Ito ↑GOF mechanism103.
The portion of SEMA3A is analogous to the Hanatoxin toxin, which binds to and inhibits potassium channels. ↑GOF of Kv4.3 potassium channels in the heart can lead to a BrS phenotype that may be associated with SCD.
Boczek et al., 103 identified a novel function for SEMA3A as a Kv4.3-specific channel blocker. Specifically, SEMA3A reduces Kv4.3 channel current density in a dose dependent manner, alters Kv4.3 channel kinetics, yet has no effect on other cardiac ion channels. SEMA3A co-immunoprecipitated with Kv4.3, suggesting a direct binding interaction between these two proteins. With the identification of rare SEMA3A mutations, leading to an overall ↑GOF in Kv4.3 current, genetic perturbations in SEMA3A may contribute to BrS103.
BrS-20 BRUGADA SYNDROME 20; BRGDA20
Locus: 1P36.3; OMIM: 601142; Gene: KCNAB2; Protein: voltage-gated K(+) channel β2-subunit (Kvβ2-R12Q) subfamily A; Proportion of BrS attributed to this genetic variants: <1% (rare). Dysfunction in the KCNAB2, which encodes the voltage-gated potassium channel β2-subunit, was associated with increased Ito activity and identified as a putative gene involved in BrS. Kvβ2 dysfunction can contribute to the Brugada ECG pattern3.
Voltage-gated potassium (Kv) channels represent the most complex class of voltage-gated ion channels from both functional and structural standpoints. Their diverse functions include regulation of neurotransmitter release, heart rate, insulin secretion, neuronal excitability, epithelial electrolyte transport, smooth muscle contraction, and cell volume. Four sequence-related potassium channel genes - shaker, shaw, shab, and shal - have been identified in Drosophila, and each has been shown to have human homolog(s).
This gene encodes a member of the potassium channel, voltage-gated, shaker-related subfamily. This member is one of the β subunits, which are auxiliary proteins associating with functional Kv-α subunits. This member alters functional properties of the KCNA4 gene product. Alternative splicing of this gene results in multiple transcript variants encoding distinct isoforms.
Portero et al., 3 combined whole-exome sequencing and linkage analysis to identify the genetic variant likely causing BrS in a pedigree for which SCN5A mutations had been excluded. This approach identified six genetic variants cosegregating with the Brugada ECG pattern within the pedigree. In silico gene prioritization pointed to one variant residing in KCNAB2, which encodes the voltage-gated K(+) channel β2-subunit (Kvβ2-R12Q)3.
Kvβ2 is widely expressed in the human heart and has been shown to interact with the fast Ito channel subunit Kv4.3, increasing its current density. By targeted sequencing of the KCNAB2 gene in 167 unrelated patients with BrS, the authors found two additional rare missense variants (L13F and V114I). They then investigated the physiological effects of the three KCNAB2 variants by using cellular electrophysiology and biochemistry3.
Patch-clamp experiments performed in COS-7 cells expressing both Kv4.3 and Kvβ2 revealed a significant increase in the current density in presence of the R12Q and L13F Kvβ2 mutants. Although biotinylation assays showed no differences in the expression of Kv4.3, the total and submembrane expression of Kvβ2-R12Q were significantly increased in comparison with wild-type Kvβ2. Altogether, their results indicate that Kvβ2 dysfunction can contribute to the Brugada ECG pattern3.
BRGDA number?
Locus: 3p25.1; OMIM: *612048; Gene: Telethonin (TCAP) TITIN-CAP. This gene belongs to the TMEM43 family104; Protein: TMEM43 transmembrane protein 43. A missense mutation, c.1073C>T (p.S358L) in the transmembrane protein 43 (TMEM43) are the cause of familial Arrhythmogenic Right Ventricular Dysplasia (ARVD) type 5 (ARVD5), also known as Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) type 5 (ARVC5)105, currently named AC.
It is an inherited disorder, often involving both ventricles, and is characterized by VT, heart failure, SCD, and fibrofatty replacement of cardiomyocytes.
This gene contains a response element for the Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) (an adipogenic transcription factor), which may explain the fibrofatty replacement of the myocardium, a characteristic pathological finding in AC. Using a positional cloning approach in a study of 15 families with ARVD/C mapping to chromosome 3 (ARVD5; 604400)106.
Merner et al.,106 identified TMEM43 as the gene mutated in this disorder. A very rare mutation in TMEM43 for the development of AC has a definite connection with desmosomal proteins (plakoglobin)107 and results in a highly arrhythmogenic form of the disease with need for ICD implantation in all male patients and in a significant number of female patients.
BrS: number?
Locus: 12q15. By genomic sequence analysis, Nakane et al. (2004) mapped the Leucine-rich repeat containing 10 (LRRC10) gene.
They mapped the mouse Lrrc10 gene to chromosome 15108; OMIM: *610846; Gene: LRRC10. Together with Receptor Accessory Protein 5, LRRC10 is a Novel Regulators of Cardiac Excitation-Contraction Coupling Structure and Function109; Ion channel and effect: INa+↓LOF; Protein: LRRC10 that is a cardiac-specific protein exclusively expressed in embryonic and adult cardiomyocytes110.
The Proportion of BrS attributed to this genetic variant: Unknown %. Phenotypes: MGI Mutant phenotypes for LRRC10: inferred from 2 alleles cardiovascular system. The first report of genetic screening of LRRC10 was published in 2016 in Chinese SUNDS victims and BrS patients. LRRC10 may be a new susceptible gene for SUNDS, and an LRRC10 variant was initially and genetically linked to BrS-associated arrhythmia111.
Silico-predicted malignant LRRC10 mutation p.E129K was detected in one SUNDS victim without pathogenic rare variants in a panel of 80 arrhythmia/cardiomyopathy-related genes. It was also shown that the rare variant p.P69L might contribute to the genetic cause for one SUNDS victim and two BrS family members. This was the first report of genetic screening of LRRC10 in Chinese SUNDS victims and BrS patients.
LRRC10 may be a new susceptible gene for SUNDS, and the LRRC10 variant was initially and genetically linked to BrS-associated arrhythmia111.
Other phenotypes: human idiopathic DCM112 and Anomalous Left Coronary Artery From The Pulmonary Artery. Gene Ontology annotations related to this gene include actin binding and alpha-actinin binding. Additionally, behavior/neurological phenotype mortality/aging muscle phenotype.
Mutations in LTCC genes, including CACNA1C, CACNA1D, CACNB2 and CACNA2D, will induce dysfunctions of calcium channels, which result in the abnormal excitations of cardiomyocytes, and finally lead to cardiac arrhythmias. Nevertheless, the newly found mutations in LTCC and their functions are continuously being elucidated. These mutations are associated with long QT syndromes, Timothy syndrome (TS, OMIM 601005)114, Brugada syndromes 3 BRGDA3, OMIM 611875)115, overlap hypertrophic cardiomyopathy, SQTSs, ERP116, CACNA1C cardiac arrhythmias, and a variety of neuropsychiatric disorders (bipolar disorder, major depression, schizophrenia, autism spectrum disorder, psychotic manifestations)124.
SCN5A mutations phenotypes: Cardiac Phenotypes: Long QT Syndrome Type 3, Brugada Syndrome, Progressive Cardiac Conduction Disease (PCCD) or Lenègre Disease; Idiopathic Ventricular Fibrillation; Early Repolarization Syndrome (ERS); Sick Sinus Syndrome (SSS); Sudden Unexplained Nocturnal Death Syndrome (SUNDS); Multifocal Ectopic Purkinje-Related Premature Contractions (MEPPE); SIDS; Overlapping Syndromes (OSs); Dilated Cardiomyopathy (DCM); Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC); Left Ventricular Noncompaction (LVNC)117; Familial Atrial Fibrillation (FAF); Gastrointestinal: Irritable Bowel Syndrome.
The cardiac potassium channels118
Cardiac potassium channels are membrane-spanning proteins that allow the passive movement of potassium ions across the cell membrane along its electrochemical gradient. They regulate the resting membrane potential, the frequency of pacemaker cells and the shape and duration of the cardiac APl. Normal potassium channel function is essential to maintain electrical stability in the heart. Gene mutations that alter the assembly, trafficking, turnover or gating of cardiac potassium channels can cause LQTS, SQTS, J-wave syndromes and AF.
1. Delayed Rectifier Potassium Currents/Channels
a) Rapid delayed rectifier potassium current (IKr) or The rapidly activating component of the delayed rectifier potassium current, IKr, rapid-rates of activation onset. Name: Kv11.1 (HERG), Gene: KCNH2, Human Chromosomal location:7q35-36 IKr and INaL are counterbalancing currents during the physiological ventricular AP and their integrals covary in individual myocytes. Targeting these ionic currents to normalize their balance may have significant therapeutic potential in heart diseases with repolarization abnormalities119.
b) The slowly activating component of the delayed rectifier potassium current, IKs, slow-rates of activation onset: Name: Kv7.1 (KVLQT1), Gene: KCNQ1. Human Chromosomal location 11p15.5. A weaker IKs contributes to the longer action potential of the M cell120.
c) The ultrarapid (IKur) ultra-rapid rates of activation onset. Name: KCNA5, Gene:12p13.3; Human chromosomal location: 12p13.3. In humans, chronic AF decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially in both atria and increases the slow component of the delayed rectifier current in both121.
2. Inward rectifying potassium channels
a) IK1, "The transient outward current". Name: Kir2.1 (IRK1); Gene: KCNJ2; Human chromosomal location: 17q23.1-24.2.
b) IKATP ATP-sensitive potassium channels, KATP. Name: Kir6.2 (BIR); Gene: KCNJ11; Human chromosomal location:11p15.1.
c) IKAch: The acetylcholine-activated potassium current, IKAch. Name: Kir3.1 (GIRK1); Gene: KCNJ3; Human chromosomal location: 2q24.111p15.1.
Inward rectifier potassium (Kir) channels typically conduct larger inward currents than outward currents, resulting in an inwardly rectifying current versus voltage relationship.
This property of inward rectification results from the voltage-dependent block of the channels by intracellular polyvalent cations and makes these channels uniquely designed for maintaining the resting potential near the potassium equilibrium potential (EK). The Kir family of channels consist of seven subfamilies of channels (Kir1.x through Kir7.x) that include the classic inward rectifier (Kir2.x) channel, the G-protein-gated inward rectifier potassium (GIRK) (Kir3.x), and the adenosine triphosphate (ATP)-sensitive (KATP) (Kir 6.x) channels as well as the renal Kir1.1 (ROMK), Kir4.1, and Kir7.1 channels.
These channels not only function to regulate electrical/electrolyte transport activity, but also serve as effector molecules for G-protein-coupled receptors (GPCRs) and as molecular sensors for cell metabolism. Of significance, Kir channels represent promising pharmacological targets for treating a number of clinical conditions, including cardiac arrhythmias, anxiety, chronic pain, and hypertension122,123.
Transient outward currents
d) Itof Ito1 4-aminopyridine (4-AP)-sensitive, calcium-independent potassium current (Ito1) is rapidly activated and inactivated in response to depolarization
e) Ito2 Ito2 4-AP-insensitive, Ca2+-activated Cl− or potassium current (Ito2)
3. Intracellular cation activated currents
a) IKNa,
b) IKCa and at least one
c) "background leak" current (IKleak)
Thus, we believe that this in-depth analytical study of the numerous mutations attributed to BrS can constitute a truly cornerstone that will help to better understand this intriguing syndrome.
CONCLUSION
- In less than three decades since the discovery of Brugada syndrome, the concept of Mendelian heredity has fallen apart. There is no doubt that the entity is oligogenetic associated with environmental factors.
- us that variants of uncertain significance, especially the rare variants of the SCN5A mutation, with European or Japanese ancestors, as well as spontaneous type 1 pattern or induced, thanks to gnomAD (coalition of researchers who seek to aggregate and harmonize exome and genome sequencing data from a variety of large-scale sequencing projects and make summary data available to the scientific community in general).
- The enormous variants and mutations found mean that we are still far from being able to concretely clarify a genotype-phenotype relationship.
REFERENCES
1.Monasky MM, Micaglio E, Ciconte G, Pappone C. Brugada syndrome: oligogenic or mendelian disease? International Journal of Molecular Sciences. janeiro de 2020; 21(5): 1687. [ Links ]
2.Wang Q, Ohno S, Ding W-G, Fukuyama M, Miyamoto A, Itoh H, et al. Gain-of-function kcnh2 mutations in patients with brugada syndrome: novel kcnh2 mutations in brugada syndrome. J Cardiovasc Electrophysiol. maio de 2014; 25(5): 522-30. [ Links ]
3.Portero V, Le Scouarnec S, Es-Salah-Lamoureux Z, Burel S, Gourraud J, Bonnaud S, et al. Dysfunction of the voltage-gated k + channel β2 subunit in a familial case of brugada syndrome. JAHA [Internet]. 13 de junho de 2016 [citado 26 de fevereiro de 2021]; 5(6). Disponível em: https://www.ahajournals.org/doi/10.1161/JAHA.115.003122 [ Links ]
4.Chen C-YJ, Lu T-P, Lin L-Y, Liu Y-B, Ho L-T, Huang H-C, et al. Impact of ancestral differences and reassessment of the classification of previously reported pathogenic variants in patients with brugada syndrome in the genomic era: a sads-tw brs registry. Front Genet. 4 de janeiro de 2019; 9:680. [ Links ]
5.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences. 1o de dezembro de 1977; 74(12): 5463-7. [ Links ]
6.Jimmy Juang J-M, Liu Y-B, Julius Chen C-Y, Yu Q-Y, Chattopadhyay A, Lin L-Y, et al. Validation and disease risk assessment of previously reported genome-wide genetic variants associated with brugada syndrome: sads-tw brs registry. Circ: Genomic and Precision Medicine [Internet]. agosto de 2020 [citado 26 de fevereiro de 2021]; 13(4). Disponível em: https://www.ahajournals.org/doi/10.1161/CIRCGEN.119.002797 [ Links ]
7.Chen CJ, Chuang EY. The puzzle of genetics in Brugada syndrome: a disease with a high risk of sudden cardiac death in young people. Ann Palliat Med. novembro de 2020; 9(6): 4394-7. [ Links ]
8.Chen C-YJ, Juang J-MJ, Lin L-Y, Liu Y-B, Ho L-T, Yu C-C, et al. Gender difference in clinical and genetic characteristics of Brugada syndrome: SADS-TW BrS registry. QJM: An International Journal of Medicine. 1o de maio de 2019;112(5):343-50. [ Links ]
9.Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. setembro de 2009; 461(7261): 272-6. [ Links ]
10.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. janeiro de 2010; 42(1): 30-5. [ Links ]
11.Wang DG. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 15 de maio de 1998; 280(5366): 1077-82. [ Links ]
12.Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Human Molecular Genetics. 15 de outubro de 2010; 19(R2): R145-51. [ Links ]
13.Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet. 1o de outubro de 2006; 140A (19): 2063-74. [ Links ]
14.Mooney SD. Progress towards the integration of pharmacogenomics in practice. Hum Genet. maio de 2015; 134(5): 459-65. [ Links ]
15.on behalf of the ESHG Public and Professional Policy Committee, van El CG, Cornel MC, Borry P, Hastings RJ, Fellmann F, et al. Whole-genome sequencing in health care: recommendations of the european society of human genetics. Eur J Hum Genet. junho de 2013; 21(6): 580-4. [ Links ]
16.Nantes Referral Center for inherited cardiac arrhythmia, Walsh R, Lahrouchi N, Tadros R, Kyndt F, Glinge C, et al. Enhancing rare variant interpretation in inherited arrhythmias through quantitative analysis of consortium disease cohorts and population controls. Genet Med. janeiro de 2021; 23(1): 47-58. [ Links ]
17.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. março de 1998; 392(6673): 293-6. [ Links ]
18.Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, et al. Reappraisal of reported genes for sudden arrhythmic death: evidence-based evaluation of gene validity for brugada syndrome. Circulation. 18 de setembro de 2018; 138(12): 1195-205. [ Links ]
19.Pérez-Riera AR, Raimundo RD, Watanabe RA, Figueiredo JL, Abreu LC de. Cardiac sodium channel, its mutations and their spectrum of arrhythmia phenotypes. J Hum Growth Dev. 28 de novembro de 2016; 26(3): 281. [ Links ]
20.Bezzina C, Veldkamp MW, van den Berg MP, Postma AV, Rook MB, Viersma J-W, et al. A single na + channel mutation causing both long-qt and brugada syndromes. Circulation Research. 3 de dezembro de 1999; 85(12): 1206-13. [ Links ]
21.Clancy CE, Tateyama M, Kass RS. Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long QT-3 syndrome. J Clin Invest. 1o de novembro de 2002; 110(9): 1251-62. [ Links ]
22.Rivolta I, Abriel H, Tateyama M, Liu H, Memmi M, Vardas P, et al. Inherited brugada and long qt-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. Journal of Biological Chemistry. agosto de 2001; 276(33): 30623-30. [ Links ]
23.Doisne N, Waldmann V, Redheuil A, Waintraub X, Fressart V, Ader F, et al. A novel gain-of-function mutation in SCN5A responsible for multifocal ectopic Purkinje-related premature contractions. Human Mutation. abril de 2020; 41(4): 850-9. [ Links ]
24.Kyndt F, Probst V, Potet F, Demolombe S, Chevallier J-C, Baro I, et al. Novel scn5a mutation leading either to isolated cardiac conduction defect or brugada syndrome in a large french family. Circulation. 17 de dezembro de 2001; 104(25): 3081-6. [ Links ]
25.Wilde AAM, Amin AS. Clinical spectrum of scn5a mutations. JACC: Clinical Electrophysiology. maio de 2018; 4(5): 569-79. [ Links ]
26.Amin AS, Boink GJJ, Atrafi F, Spanjaart AM, Asghari-Roodsari A, Molenaar RJ, et al. Facilitatory and inhibitory effects of SCN5A mutations on atrial fibrillation in Brugada syndrome. Europace. 1o de julho de 2011; 13(7): 968-75. [ Links ]
27.Han D, Tan H, Sun C, Li G. Dysfunctional Nav1.5 channels due to SCN5A mutations. Exp Biol Med (Maywood). junho de 2018; 243(10): 852-63. [ Links ]
28.Remme CA, Wilde AAM, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of scn5a mutations. Trends in Cardiovascular Medicine. abril de 2008; 18(3): 78-87. [ Links ]
29.Wilde AAM, Garan H, Boyden PA. Role of the Purkinje system in heritable arrhythmias. Heart Rhythm. julho de 2019; 16(7): 1121-6. [ Links ]
30.Haïssaguerre M, Duchateau J, Dubois R, Hocini M, Cheniti G, Sacher F, et al. Idiopathic ventricular fibrillation. JACC: Clinical Electrophysiology. junho de 2020; 6(6): 591-608. [ Links ]
31.Robyns T, Nuyens D, Vandenberk B, Kuiperi C, Corveleyn A, Breckpot J, et al. Genotype-phenotype relationship and risk stratification in loss-of-function SCN 5A mutation carriers. Ann Noninvasive Electrocardiol [Internet]. setembro de 2018 [citado 26 de fevereiro de 2021]; 23(5). Disponível em: https://onlinelibrary.wiley.com/doi/abs/10.1111/anec.12548 [ Links ]
32.Amin AS, Reckman YJ, Arbelo E, Spanjaart AM, Postema PG, Tadros R, et al. SCN5A mutation type and topology are associated with the risk of ventricular arrhythmia by sodium channel blockers. International Journal of Cardiology. setembro de 2018; 266: 128-32. [ Links ]
33.Rao AS, Knowles JW. Polygenic risk scores in coronary artery disease. Current Opinion in Cardiology. julho de 2019; 34(4): 435-40. [ Links ]
34.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. setembro de 2018; 50(9): 1219-24. [ Links ]
35.Micaglio E, Monasky MM, Ciconte G, Vicedomini G, Conti M, Mecarocci V, et al. Novel scn5a frameshift mutation in brugada syndrome associated with complex arrhythmic phenotype. Front Genet. 6 de junho de 2019; 10: 547. [ Links ]
36.Monasky M, Micaglio E, Giachino D, Ciconte G, Giannelli L, Locati E, et al. Genotype-phenotype correlation in a family with brugada syndrome harboring the novel p. Gln371* nonsense variant in the scn5a gene. IJMS. 6 de novembro de 2019; 20(22): 5522. [ Links ]
37.Monasky MM, Micaglio E, Ciconte G, Benedetti S, Di Resta C, Vicedomini G, et al. Genotype/phenotype relationship in a consanguineal family with brugada syndrome harboring the r1632c missense variant in the scn5a gene. Front Physiol. 28 de maio de 2019; 10: 666. [ Links ]
38.Micaglio E, Monasky MM, Ciconte G, Vicedomini G, Conti M, Mecarocci V, et al. Scn5a nonsense mutation and nf1 frameshift mutation in a family with brugada syndrome and neurofibromatosis. Front Genet. 15 de fevereiro de 2019; 10: 50. [ Links ]
39.Micaglio E, Monasky M, Resta N, Bagnulo R, Ciconte G, Giannelli L, et al. Novel scn5a p. W697x nonsense mutation segregation in a family with brugada syndrome. IJMS. 4 de outubro de 2019; 20(19): 4920. [ Links ]
40.Veltmann C, Barajas-Martinez H, Wolpert C, Borggrefe M, Schimpf R, Pfeiffer R, et al. Further insights in the most common scn5a mutation causing overlapping phenotype of long qt syndrome, brugada syndrome, and conduction defect. JAHA [Internet]. 6 de julho de 2016 [citado 26 de fevereiro de 2021]; 5(7). Disponível em: https://www.ahajournals.org/doi/10.1161/JAHA.116.003379 [ Links ]
41.Samani K, Ai T, Towbin JA, Brugada R, Shuraih M, Xi Y, et al. A nonsense scn5a mutation associated with brugada-type electrocardiogram and intraventricular conduction defects. Pacing and Clinical Electrophysiology. setembro de 2009; 32(9): 1231-6. [ Links ]
42.Probst V, Allouis M, Sacher F, Pattier S, Babuty D, Mabo P, et al. Progressive cardiac conduction defect is the prevailing phenotype in carriers of a brugada syndrome scn5a mutation. J Cardiovasc Electrophysiol. março de 2006; 17(3): 270-5. [ Links ]
43.Keller DI, Barrane FZ, Gouas L, Martin J, Pilote S, Suarez V, Osswald S, Brink M, Guicheney P, Schwick N, Chahine M. A novel nonsense mutation in the SCN5A gene leads to Brugada syndrome and a silent gene mutation carrier state. Can J Cardiol. 2005; 21: 925-931. [ Links ]
44.Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, Timothy KW, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: Implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. novembro de 2004; 1(5): 600-7. [ Links ]
45.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. janeiro de 2010; 7(1): 33-46. [ Links ]
46.Ohkubo K, Watanabe I, Okumura Y, Ashino S, Kofune M, Nagashima K, et al. Prolonged qrs duration in lead v2 and risk of life-threatening ventricular arrhythmia in patients with brugada syndrome. Int Heart J. 2011; 52(2): 98-102. [ Links ]
47.Nademanee K, Raju H, de Noronha SV, Papadakis M, Robinson L, Rothery S, et al. Fibrosis, connexin-43, and conduction abnormalities in the brugada syndrome. Journal of the American College of Cardiology. novembro de 2015; 66(18): 1976-86. [ Links ]
48.Ingles J, Macciocca I, Morales A, Thomson K. Genetic testing in inherited heart diseases. Heart, Lung and Circulation. abril de 2020; 29(4): 505-11. [ Links ]
49.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, et al. Mutation in glycerol-3-phosphate dehydrogenase 1-like gene (gpd1-l) decreases cardiac na + current and causes inherited arrhythmias. Circulation. 13 de novembro de 2007; 116(20): 2260-8. [ Links ]
50.Antzelevitch C, Patocskai B. Brugada syndrome: clinical, genetic, molecular, cellular, and ionic aspects. Current Problems in Cardiology. janeiro de 2016; 41(1): 7-57. [ Links ]
51.Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1-like gene (gpd1-l) mutations in sudden infant death syndrome. Circulation. 13 de novembro de 2007; 116(20): 2253-9. [ Links ]
52.Béziau DM, Barc J, O'Hara T, Le Gloan L, Amarouch MY, Solnon A, et al. Complex Brugada syndrome inheritance in a family harbouring compound SCN5A and CACNA1C mutations. Basic Res Cardiol. novembro de 2014; 109(6): 446. [ Links ]
53.Bozarth X, Dines JN, Cong Q, Mirzaa GM, Foss K, Lawrence Merritt J, et al. Expanding clinical phenotype in CACNA1C related disorders: From neonatal onset severe epileptic encephalopathy to late-onset epilepsy. Am J Med Genet. dezembro de 2018; 176(12): 2733-9. [ Links ]
54.Ou X, Crane DE, MacIntosh BJ, Young LT, Arnold P, Ameis S, et al. CACNA1C rs1006737 genotype and bipolar disorder: Focus on intermediate phenotypes and cardiovascular comorbidity. Neuroscience & Biobehavioral Reviews. agosto de 2015; 55: 198-210. [ Links ]
55.Tesli M, Skatun KC, Ousdal OT, Brown AA, Thoresen C, Agartz I, et al. Cacna1c risk variant and amygdala activity in bipolar disorder, schizophrenia and healthy controls. Fatemi H, organizador. PLoS ONE. 20 de fevereiro de 2013; 8(2): e56970. [ Links ]
56.Chernyavskaya Y, Ebert AM, Milligan E, Garrity DM. Voltage-gated calcium channel CACNB2 (β2.1) protein is required in the heart for control of cell proliferation and heart tube integrity. Dev Dyn. abril de 2012; 241(4): 648-62. [ Links ]
57.Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Phillips HA, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel ß1 subunit gene SCN1B. Nat Genet. agosto de 1998; 19(4): 366-70. [ Links ]
58.Singh R, Andermann E, Whitehouse WPA, Harvey AS, Keene DL, Seni M-H, et al. Severe myoclonic epilepsy of infancy: extended spectrum of gefs+? Epilepsia. 20 de dezembro de 2001; 42(7): 837-44. [ Links ]
59.Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, et al. Mutations in sodium channel β1- and β2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. junho de 2009; 2(3): 268-75. [ Links ]
60.Scheffer IE, Harkin LA, Grinton BE, Dibbens LM, Turner SJ, Zielinski MA, et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain. 21 de novembro de 2006; 130(1): 100-9. [ Links ]
61.Ogiwara I, Nakayama T, Yamagata T, Ohtani H, Mazaki E, Tsuchiya S, et al. A homozygous mutation of voltage-gated sodium channel β I gene SCN1B in a patient with Dravet syndrome: Homozygous SCN1B Mutation in Epilepsy. Epilepsia. dezembro de 2012; 53(12): e200-3. [ Links ]
62.Delpón E, Cordeiro JM, Núñez L, Thomsen PEB, Guerchicoff A, Pollevick GD, et al. Functional effects of kcne3 mutation and its role in the development of brugada syndrome. Circ Arrhythm Electrophysiol. agosto de 2008; 1(3): 209-18. [ Links ]
63.Nakajima T, Wu J, Kaneko Y, Ashihara T, Ohno S, Irie T, et al. Kcne3 t4a as the genetic basis of brugada-pattern electrocardiogram. Circ J. 2012; 76(12): 2763-72. [ Links ]
64.Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, et al. A mutation in the β3 subunit of the cardiac sodium channel associated with brugada ecg phenotype. Circ Cardiovasc Genet. junho de 2009; 2(3): 270-8. [ Links ]
65.Nielsen MW, Holst AG, Olesen S-P, Olesen MS. The genetic component of Brugada syndrome. Front Physiol [Internet]. 2013 [citado 26 de fevereiro de 2021]; 4. Disponível em: http://journal.frontiersin.org/article/10.3389/fphys.2013.00179/abstract [ Links ]
66.Ishikawa T, Takahashi N, Ohno S, Sakurada H, Nakamura K, On YK, et al. Novel scn3b mutation associated with brugada syndrome affects intracellular trafficking and function of nav1. 5. Circ J. 2013; 77(4): 959-67. [ Links ]
67.Ueda K, Hirano Y, Higashiuesato Y, Aizawa Y, Hayashi T, Inagaki N, et al. Role of HCN4 channel in preventing ventricular arrhythmia. J Hum Genet. fevereiro de 2009; 54(2): 115-21. [ Links ]
68.Barajas-Martínez H, Hu D, Ferrer T, Onetti CG, Wu Y, Burashnikov E, et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm. abril de 2012; 9(4): 548-55. [ Links ]
69.Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, et al. Transient outward current (Ito) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. julho de 2011; 8(7): 1024-32. [ Links ]
70.Templin C, Ghadri J-R, Rougier J-S, Baumer A, Kaplan V, Albesa M, et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (Sqts6). European Heart Journal. 1o de maio de 2011;3 2(9): 1077-88. [ Links ]
71.Hancox JC, Whittaker DG, Du C, Stuart AG, Zhang H. Emerging therapeutic targets in the short QT syndrome. Expert Opinion on Therapeutic Targets. 4 de maio de 2018; 22(5): 439-51. [ Links ]
72.Iles DE, Lehmann-Horn F, Scherer SW, Tsui L-C, Weghuis DO, Suijkerbuijk RF, et al. Localization of the gene encoding the α 2 /δ-subunits of the L-type voltage-dependent calcium channel to chromosome 7q and analysis of the segregation of flanking markers in malignant hyperthermia susceptible families. Hum Mol Genet. 1994; 3(6): 969-75. [ Links ]
73.Portero V, Wilders R, Casini S, Charpentier F, Verkerk AO, Remme CA. Kv4. 3 expression modulates nav1. 5 sodium current. Front Physiol. 12 de março de 2018; 9: 178. [ Links ]
74.Campuzano O, Berne P, Selga E, Allegue C, Iglesias A, Brugada J, Brugada R. Brugada syndrome and p.E61X_RANGRF. Cardiol J. 2014; 21: 121-127. [ Links ]
75.Olesen MS, Jensen NF, Holst AG, Nielsen JB, Tfelt-Hansen J, Jespersen T, et al. A novel nonsense variant in nav1. 5 cofactor mog1 eliminates its sodium current increasing effect and may increase the risk of arrhythmias. Canadian Journal of Cardiology. julho de 2011; 27(4): 523.e17-523.e23. [ Links ]
76.Wu L, Yong SL, Fan C, Ni Y, Yoo S, Zhang T, et al. Identification of a new co-factor, mog1, required for the full function of cardiac sodium channel nav1. 5. Journal of Biological Chemistry. março de 2008; 283(11): 6968-78. [ Links ]
77.Chakrabarti S, Wu X, Yang Z, Wu L, Yong SL, Zhang C, et al. Mog1 rescues defective trafficking of na v 1. 5 mutations in brugada syndrome and sick sinus syndrome. Circ Arrhythm Electrophysiol. abril de 2013; 6(2): 392-401. [ Links ]
78.Mlynarova J, Trentin-Sonoda M, Gaisler da Silva F, Major JL, Salih M, Carneiro-Ramos MS, et al. SLMAP3 isoform modulates cardiac gene expression and function. Backx PH, organizador. PLoS ONE. 1o de abril de 2019; 14(4): e0214669. [ Links ]
79.Ishikawa T, Sato A, Marcou CA, Tester DJ, Ackerman MJ, Crotti L, et al. A novel disease gene for brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hnav1. 5. Circ Arrhythm Electrophysiol. dezembro de 2012; 5(6): 1098-107. [ Links ]
80.Hu D, Barajas-Martínez H, Terzic A, Park S, Pfeiffer R, Burashnikov E, et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. International Journal of Cardiology. fevereiro de 2014; 171(3): 431-42. [ Links ]
81.Riuró H, Beltran-Alvarez P, Tarradas A, Selga E, Campuzano O, Vergés M, et al. A missense mutation in the sodium channel β2 subunit reveals scn2b as a new candidate gene for brugada syndrome. Human Mutation. julho de 2013; 34(7): 961-6. [ Links ]
82.Bao Y, Willis BC, Frasier CR, Lopez-Santiago LF, Lin X, Ramos-Mondragón R, et al. scn2b deletion in mice results in ventricular and atrial arrhythmias. Circ Arrhythm Electrophysiol [Internet]. dezembro de 2016 [citado 26 de fevereiro de 2021]; 9(12). Disponível em: https://www.ahajournals.org/doi/10.1161/CIRCEP.116.003923 [ Links ]
83.Huang L, Tang S, Peng L, Chen Y, Cheng J. Molecular autopsy of desmosomal protein plakophilin-2 in sudden unexplained nocturnal death syndrome. J Forensic Sci. maio de 2016; 61(3): 687-91. [ Links ]
84.Cerrone M, Delmar M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends in Cardiovascular Medicine. julho de 2014; 24(5): 184-90. [ Links ]
85.Papavassiliu T, Wolpert C, Flüchter S, Schimpf R, Neff W, Haase KK, et al. Magnetic resonance imaging findings in patients with brugada syndrome. Journal of Cardiovascular Electrophysiology. outubro de 2004; 15(10): 1133-8. [ Links ]
86.Cerrone M, Noorman M, Lin X, Chkourko H, Liang F-X, van der Nagel R, et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovascular Research. 1o de setembro de 2012; 95(4): 460-8. [ Links ]
87.Wang C, Wang C, Hoch EG, Pitt GS. Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage-gated sodium channels. Journal of Biological Chemistry. julho de 2011; 286(27): 24253-63. [ Links ]
88.Hennessey JA, Marcou CA, Wang C, Wei EQ, Wang C, Tester DJ, et al. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm. dezembro de 2013; 10(12): 1886-94. [ Links ]
89.Villeneuve N, Abidi A, Cacciagli P, Mignon-Ravix C, Chabrol B, Villard L, et al. Heterogeneity of FHF1 related phenotype: Novel case with early onset severe attacks of apnea, partial mitochondrial respiratory chain complex II deficiency, neonatal onset seizures without neurodegeneration. European Journal of Paediatric Neurology. setembro de 2017; 21(5): 783-6. [ Links ]
90.Hu D, Barajas-Martínez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, et al. Mutations in scn10a are responsible for a large fraction of cases of brugada syndrome. Journal of the American College of Cardiology. julho de 2014; 64(1): 66-79. [ Links ]
91.Sotoodehnia N, Isaacs A, de Bakker PIW, Dörr M, Newton-Cheh C, Nolte IM, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. dezembro de 2010; (12): 1068-76. [ Links ]
92.Abriel H. Genetic background of Brugada syndrome is more complex than what we would like it to be! Cardiovascular Research. 1o de junho de 2015; 106(3): 351-2. [ Links ]
93.Behr ER, Savio-Galimberti E, Barc J, Holst AG, Petropoulou E, Prins BP, et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovascular Research. 1o de junho de 2015; 106(3): 520-9. [ Links ]
94.Fukuyama M, Ohno S, Makiyama T, Horie M. Novel SCN10A variants associated with Brugada syndrome. Europace. junho de 2016; 18(6): 905-11. [ Links ]
95.El-Battrawy I, Albers S, Cyganek L, Zhao Z, Lan H, Li X, et al. A cellular model of Brugada syndrome with SCN10A variants using human-induced pluripotent stem cell-derived cardiomyocytes. EP Europace. 1o de setembro de 2019; 21(9): 1410-21. [ Links ]
96.Gray B, Hasdemir C, Ingles J, Aiba T, Makita N, Probst V, et al. Lack of genotype-phenotype correlation in Brugada Syndrome and Sudden Arrhythmic Death Syndrome families with reported pathogenic SCN1B variants. Heart Rhythm. julho de 2018; 15(7): 1051-7. [ Links ]
97.Leo S, D'Hooge R, Meert T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behavioural Brain Research. março de 2010; 208(1): 149-57. [ Links ]
98.Leimeister C, Externbrink A, Klamt B, Gessler M. Hey genes: a novel subfamily of hairy - and Enhancer of split related genes specifically expressed during mouse embryogenesis. Mechanisms of Development. julho de 1999; 85(1-2): 173-7. [ Links ]
99.Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud J-B, Simonet F, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. setembro de 2013; 45(9): 1044-9. [ Links ]
100.Veerman CC, Podliesna S, Tadros R, Lodder EM, Mengarelli I, de Jonge B, et al. The brugada syndrome susceptibility gene hey2 modulates cardiac transmural ion channel patterning and electrical heterogeneity. Circ Res. 18 de agosto de 2017; 121(5): 537-48. [ Links ]
101.Andreasen L, Nielsen JB, Darkner S, Christophersen IE, Jabbari J, Refsgaard L, et al. Brugada syndrome risk loci seem protective against atrial fibrillation. Eur J Hum Genet. dezembro de 2014; 22(12): 1357-61. [ Links ]
102.Nakano Y, Ochi H, Onohara Y, Toshishige M, Tokuyama T, Matsumura H, et al. Common variant near hey2 has a protective effect on ventricular fibrillation occurrence in brugada syndrome by regulating the repolarization current. Circ Arrhythm Electrophysiol [Internet]. janeiro de 2016 [citado 26 de fevereiro de 2021]; 9(1). Disponível em: https://www.ahajournals.org/doi/10.1161/CIRCEP.115.003436 [ Links ]
103.Boczek NJ, Ye D, Johnson EK, Wang W, Crotti L, Tester DJ, et al. Characterization of sema3a -encoded semaphorin as a naturally occurring k v 4. 3 protein inhibitor and its contribution to brugada syndrome. Circ Res. agosto de 2014; 115(4): 460-9. [ Links ]
104.Turker I, Makiyama T, Ueyama T, Shimizu A, Yamakawa M, Chen P, et al. telethonin variants found in brugada syndrome, j-wave pattern ecg, and arvc reduce peak na v 1. 5 currents in hek-293 cells. Pacing Clin Electrophysiol. agosto de 2020; 43(8): 838-46. [ Links ]
105.Baskin B, Skinner JR, Sanatani S, Terespolsky D, Krahn AD, Ray PN, et al. TMEM43 mutations associated with arrhythmogenic right ventricular cardiomyopathy in non-Newfoundland populations. Hum Genet. novembro de 2013; 132(11): 1245-52. [ Links ]
106.Merner ND, Hodgkinson KA, Haywood AFM, Connors S, French VM, Drenckhahn J-D, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the tmem43 gene. The American Journal of Human Genetics. abril de 2008; 82(4): 809-21. [ Links ]
107.Siragam V, Cui X, Masse S, Ackerley C, Aafaqi S, Strandberg L, et al. Tmem43 mutation p. S358l alters intercalated disc protein expression and reduces conduction velocity in arrhythmogenic right ventricular cardiomyopathy. Ai X, organizador. PLoS ONE. 24 de outubro de 2014; 9(10): e109128. [ Links ]
108.Nakane T, Satoh T, Inada Y, Nakayama J, Itoh F, Chiba S. Molecular cloning and expression of HRLRRP, a novel heart-restricted leucine-rich repeat protein. Biochemical and Biophysical Research Communications. fevereiro de 2004; 314(4): 1086-92. [ Links ]
109.Chiamvimonvat N, Song L. Lrrc10 (Leucine-rich repeat containing protein 10) and reep5 (Receptor accessory protein 5) as novel regulators of cardiac excitation-contraction coupling structure and function. JAHA [Internet]. 6 de fevereiro de 2018 [citado 26 de fevereiro de 2021]; 7(3). Disponível em: https://www.ahajournals.org/doi/10.1161/JAHA.117.008260 [ Links ]
110.Brody MJ, Hacker TA, Patel JR, Feng L, Sadoshima J, Tevosian SG, et al. Ablation of the cardiac-specific gene leucine-rich repeat containing 10 (Lrrc10) results in dilated cardiomyopathy. Peng T, organizador. PLoS ONE. 7 de dezembro de 2012; 7(12): e51621. [ Links ]
111.Huang L, Tang S, Chen Y, Zhang L, Yin K, Wu Y, et al. Molecular pathological study on LRRC10 in sudden unexplained nocturnal death syndrome in the Chinese Han population. Int J Legal Med. maio de 2017; 131(3): 621-8. [ Links ]
112.Brody MJ, Lee Y. The role of leucine-rich repeat containing protein 10 (Lrrc10) in dilated cardiomyopathy. Front Physiol [Internet]. 3 de agosto de 2016 [citado 26 de fevereiro de 2021]; 7. Disponível em: http://journal.frontiersin.org/Article/10.3389/fphys.2016.00337/abstract [ Links ]
113.Campuzano O, Sarquella-Brugada G, Cesar S, Arbelo E, Brugada J, Brugada R. Update on genetic basis of brugada syndrome: monogenic, polygenic or oligogenic? IJMS. 28 de setembro de 2020; 21(19): 7155. [ Links ]
114.Han D, Xue X, Yan Y, Li G. Dysfunctional Cav1.2 channel in Timothy syndrome, from cell to bedside. Exp Biol Med (Maywood). setembro de 2019; 244(12): 960-71. [ Links ]
115.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by st-segment elevation, short qt intervals, and sudden cardiac death. Circulation. 30 de janeiro de 2007; 115(4): 442-9. [ Links ]
116.Chen Y, Barajas-Martinez H, Zhu D, Wang X, Chen C, Zhuang R, et al. Novel trigenic CACNA1C/DES/MYPN mutations in a family of hypertrophic cardiomyopathy with early repolarization and short QT syndrome. J Transl Med. dezembro de 2017; 15(1):7 8. [ Links ]
117.Pappone C, Monasky MM, Ciconte G. Epicardial ablation in genetic cardiomyopathies: a new frontier. European Heart Journal Supplements. 1o de março de 2019; 21(Supplement_B): B61-6. [ Links ]
118.Crotti L, Odening KE, Sanguinetti MC. Heritable arrhythmias associated with abnormal function of cardiac potassium channels. Cardiovascular Research. 15 de julho de 2020; 116(9): 1542-56. [ Links ]
119.Hegyi B, Chen-Izu Y, Izu LT, Rajamani S, Belardinelli L, Bers DM, et al. Balance between rapid delayed rectifier k + current and late na + current on ventricular repolarization: an effective antiarrhythmic target? Circ: Arrhythmia and Electrophysiology [Internet]. abril de 2020 [citado 26 de fevereiro de 2021]; 13(4). Disponível em: https://www.ahajournals.org/doi/10.1161/CIRCEP.119.008130 [ Links ]
120.Liu D-W, Antzelevitch C. Characteristics of the delayed rectifier current (I Kr and I Ks) in canine ventricular epicardial, midmyocardial, and endocardial myocytes: a weaker i ks contributes to the longer action potential of the m cell. Circulation Research. março de 1995; 76(3): 351-65. [ Links ]
121.Caballero R, de la Fuente MG, Gómez R, Barana A, Amorós I, Dolz-Gaitón P, et al. In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. Journal of the American College of Cardiology. maio de 2010; 55(21): 2346-54. [ Links ]
122.Walsh KB. Screening technologies for inward rectifier potassium channels: discovery of new blockers and activators. SLAS DISCOVERY: Advancing the Science of Drug Discovery. junho de 2020; 25(5): 420-33. [ Links ]
123.Tonussi Mendes JE, Nikus K, Barbosa-Barros R, Pérez-Riera AR. The numerous denominations of the Brugada syndrome and proposal about how to put an end to an old controversy - a historical-critical perspective. J Hum Growth Dev. 2020; 30(3): 480-491. [ Links ]
124.Pérez-Riera AR, Raimundo RD, Watanabe RA, Abreu LC. The cardiac sodium channel its mutations and their spectrum arrhythmia phenotypes. J Hum Growth Dev. 2016; 26(3): 281-296. Doi: http://dx.doi.org/10.7322/jhgd.122759 [ Links ]
 Correspondence:
Correspondence:
arperezriera@gmail.com
luizcarlos.deabreu@ul.ie
Manuscript received: December 2020
Manuscript accepted: February 2021
Version of record online: March 2021

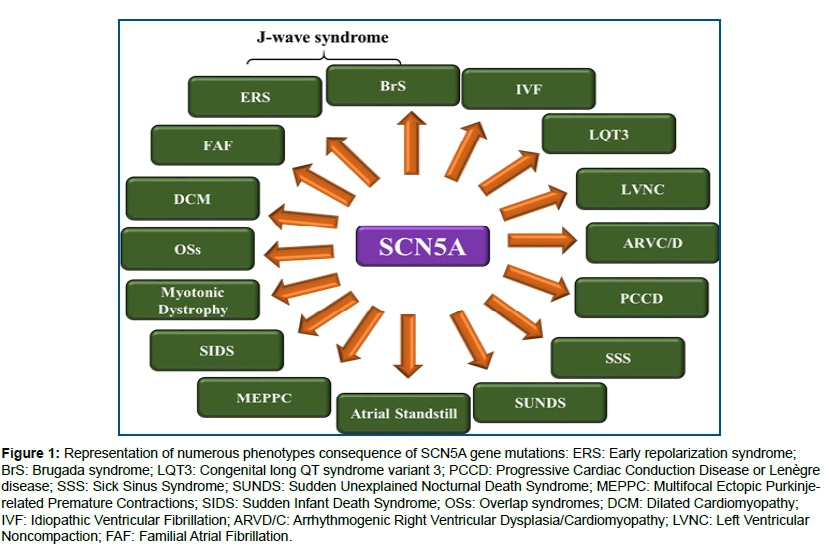{kind=link}
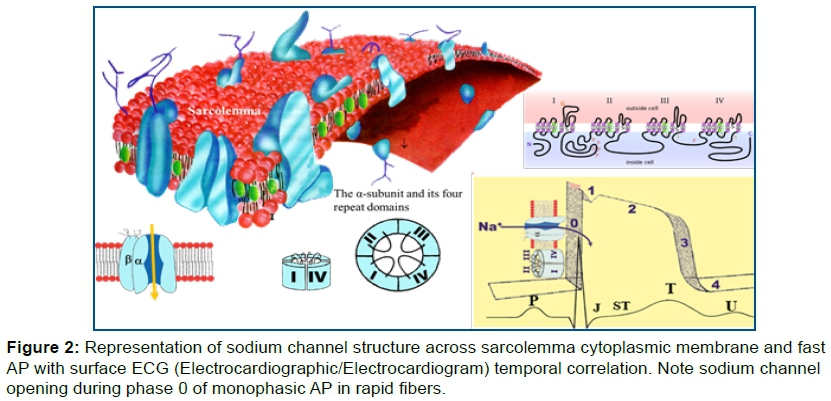{kind=link}
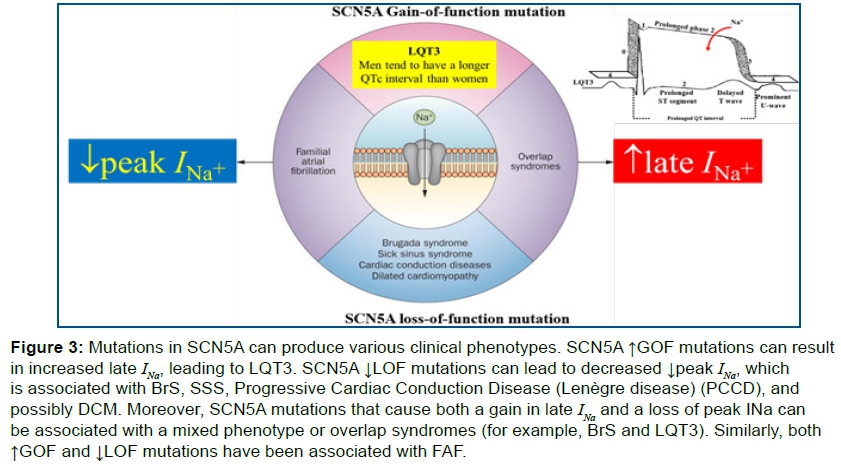{kind=link}
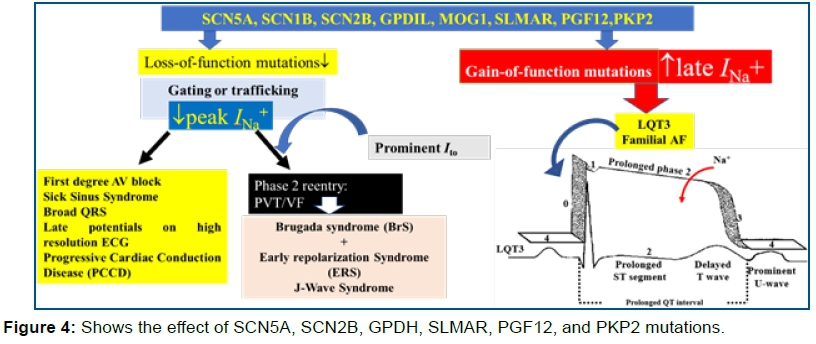{kind=link}
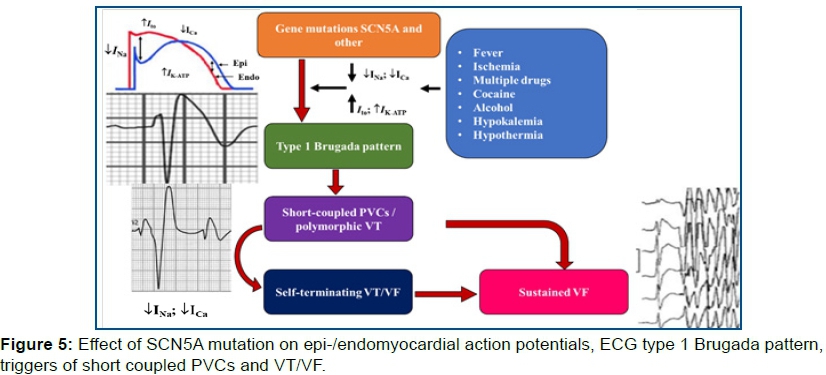{kind=link}
{kind=link}
{kind=link}
{kind=link}
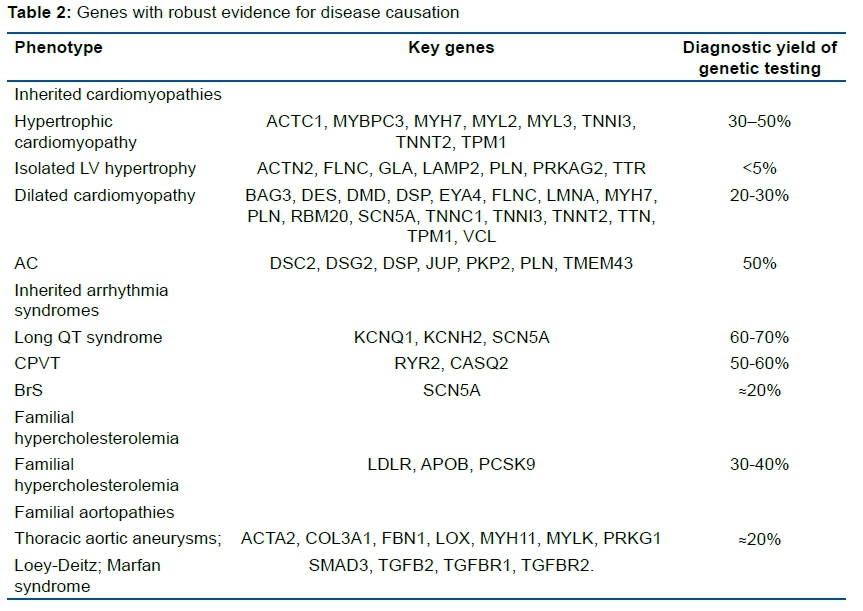{kind=link}
{kind=link}
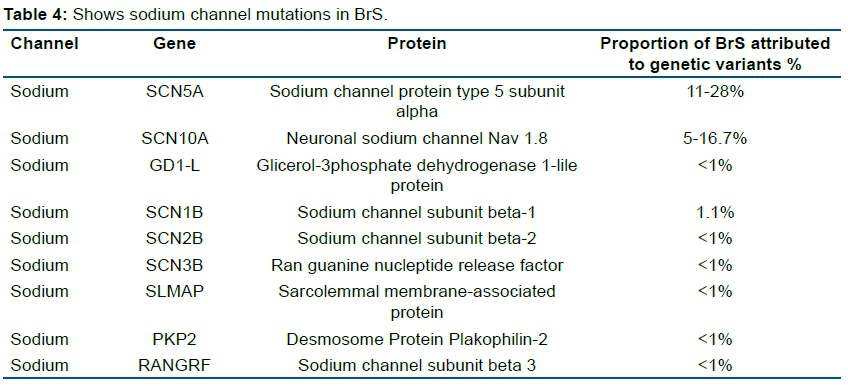{kind=link}
{kind=link}