








Serviços Personalizados
Journal

artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores
Compartilhar

 Permalink
PermalinkCadernos de Pós-Graduação em Distúrbios do Desenvolvimento
versão impressa ISSN 1519-0307versão On-line ISSN 1809-4139
Cad. Pós-Grad. Distúrb. Desenvolv. vol.18 no.1 São Paulo jan./jun. 2018
https://doi.org/10.5935/cadernosdisturbios.v18n1p148-163
Genetic and functional differences between Bethlem miopathyand Ullrich congenital muscular dystrophy - case studies
Diferenças funcionais e genéticas da miopatia de Bethlem e distrofia muscular congênita de Ullrich - estudo de casos
Diferencias genéticas y funcionales de la miopatía de Bethlem y distrofia muscular congénita de Ullrich - estudio de casos
Juliana Aparecida Rhein TellesI; Mariana Calil VoosII; Isabella Pessa AnequiniIII; Francis Meire FaveroIV; Thiago Henrique SilvaV; Fátima Aparecida CaromanoVI
IUniversidade de São Paulo (USP), São Paulo, SP, Brasil. E-mail: juliana.rhein@bol.com.br
IIUniversidade de São Paulo (USP), São Paulo, SP, Brasil. Universidade Ibirapuera, São Paulo, SP, Brasil. E-mail: ftmarivoos@gmail.com
IIIUniversidade de São Paulo (USP), São Paulo, SP, Brasil. E-mail: isa.anequini@gmail.com
IVUniversidade de São Paulo (USP), São Paulo, SP, Brasil. E-mail: ffave.nexp@latoneuro.com.br
VUniversidade de São Paulo (USP), São Paulo, SP, Brasil. E-mail: thi1822@gmail.com
VIUniversidade de São Paulo (USP), São Paulo, SP, Brasil. E-mail: fcaromano@uol.com.br
ABSTRACT
INTRODUCTION: Bethlem myopathy (BM) and Ullrich Congenital Muscular Dystrophy (DMCU) result from a mutation in collagen type VI.
OBJECTIVE: Describe the functionality of BM and UMCD subjects, at the Center for Human Genome Studies.
METHODS: It was researched functional skills in both cases, with muscle strength and shortening evaluation.
RESULTS: Muscular strength and functional losses in two cases, with a worse score in DMCU.
Keywords: Bethlem Myopathy. Ullrich Congenital Muscular. Type VI collagen. Functional Tests. Disabilities.
RESUMO
INTRODUÇÃO: A Miopatia de Bethlem (MB) e a distrofia muscular congênita de Ullrich (DMCU) são resultados de uma mutação no colágeno VI.
OBJETIVO: Descrever a funcionalidade na MB e DMCU, seguidos no Centro de Estudos do Genoma Humano.
MÉTODO: Foram pesquisadas habilidades funcionais dos dois casos, bem como força muscular e encurtamentos.
RESULTADOS: Perdas musculares e funcionais nos dois casos, com piora no caso DMCU.
Palavras-chave: Miopatia de Bethlem. Distrofia muscular congênita. Colágeno tipo VI. Testes funcionais. Incapacidade.
RESUMEN
INTRODUCCIÓN: La Miopatía de Bethlem (MB) y la Distrofia Muscular Congénita de Ullrich (DMCU) son resultados de una mutación en el colágeno VI.
METAS: Describir la funcionalidad en la MB y DMCU, seguidos en el Centro de Estudios del Genoma Humano. Se investigó habilidades funcionales de ambos y
MÉTODO: Furon investigadas habilidades funcionales de los casos, así como fuerza muscular y acortamiento.
RESULTADOS: Pérdidas de fuerza muscular y funcionale en los dos casos y un empeoramiento en DMCU
Palabras-clave: Bethlem Miopathy. Ullrich Congenital Muscular. Type VI collagen. Functional tests. Incapacidad.
INTRODUCTION
Mutations in COL6A1, COL6A2, and COL6A3, cause a congenital illness group. The type VI collagen is an extracellular protein forming a distinct myofibrillar network of most interstitial connective tissues, existing in the cellular matrices of muscle, skin, tendon, cartilage, intervertebral discs, blood vessels and eyes.
There is a role of integrity maintenance and skeletal muscle function (TAGLIAVINI et al., 2014; BONNEMANN, 2011). In the extracellular matrix, type VI collagen interacts with other components, including collagen type I, collagen type II and collagen type XIV (TAGLIAVINI et al., 2014).
The Bethlem myopathy is an autosomal dominant or recessive disorder characterized by proximal and axial progressive muscle weakness, with flexion finger contractures (TAGLIAVINI et al., 2014).
In 1976, Bethlem e van Wijngaarden described three Dutch families with 28 patients with myopathy with an autosomal dominant disorder. The initial clinical manifestations are in the childhood, with a slow progression with a part of the patients achieving the adult age. The patients present a medium weakness and atrophy of trunk and limbs muscles, with a proximal and extensors muscles most affected. Contractures in elbow flexors articulations and plantar flexion were found (BETHLEM; WIJGAARDEN, 1976).
Many characteristics are common just like muscle disability and fatigue, contractures often in fingers, elbows, and ankles. The illness course is slow in most part of patients, and the progression of muscular weakness can occur in the fifth decade of life (TAGLIAVINI et al., 2014; BONNEMANN, 2011; ZAMURS et al., 2015; TONI et al., 2014; PALMA et al., 2014).
There is considerable variability in BM phenotypes. Although the description of symptoms must be in adulthood, the clinical dysfunctions experienced by these patients may often be noted in early childhood. Affected babies may present hypotonia, deformities in the feet and congenital torticollis (in which it is noted in more than 50% of patients). However, congenital contractures tend to be resolved in the first years of life. Very young children may have mild weakness and, in this age group, present ligament laxity rather than contractures. Some individuals have proximal weakness without apparent contractures and have been diagnosed as muscular dystrophy of waists. Others have predominantly more contractures, without weakness. In some individuals, the muscles are very rigid and a specific recessive mutation (BONNEMANN, 2011).
The Ullrich congenital muscular dystrophy is a severe disorder characterized by congenital muscle weakness with axial and proximal contractures concurrently associated with distal joint hypermobility (TAGLIAVINI et al., 2014).
In 1930, Otto Ullrich described two boys with an uncommon congenital myopathy subtype caused by a mutation in homozygous and characterized for muscular weakness and fatigue, associated with distal ligament laxity and proximal contractures since birth until childhood (VOERMANS et al., 2009; REED et al., 2005; YONEKAWA; NISHINO, 2015; REED, 2009; ZOU et al., 2014).
There is a classification according to the myopathy and type VI collagen, referring to the disease symptoms and staging. There are:
• Ullrich Congenital Muscular Dystrophy severe (early severity): The patient acquires the motor ability to sit, sometimes crawl, but without independent deambulation, severe contractures may appear.
• Ullrich Congenital Muscular Dystrophy moderately Progressive (moderately evolution): Patient achieves independent deambulation, after some delay in the acquisition, but this ability is lost around 5 to 15 years, severe contractures may appear.
• Intermediate phenotype (mild): The deambulation keeps up until 20 years, and the contractures are variable (BONNEMANN, 2011).
The symptoms are presented as a severe form, in this case, the symptoms are from birth through early childhood, leading the patient to lose his independent gait.in adolescence, dying as a result of respiratory failure (ZAMURS et al., 2015; PALMA et al., 2014; YONEKAWA; NISHINO, 2015; CARAKUSHANSKY; RIBEIRO; KAHN, 2012).
The clinical manifestations of UCMD are an early muscular weakness, proximal and distal contractures, hyperextensibility and normal cognitive. Also occurs spinal stiffness, kyphoscoliosis, distal articular hyperextensibility and thinner calcaneus tendon. In the first and second decade of life, must occur respiratory failure.
In the first and second decade of life, respiratory failure may occur, and it is one of the main causes of death when not correctly treated with nocturnal ventilatory support. Some dermatological symptoms may also occur as hypertrophic scars and follicular hyperkeratosis (ROZORGMEHR et al., 2013).
Both clinical features and genetic findings, Bethlem myopathy, and Ullrich congenital muscular dystrophy can be considered similar when the end is analyzed within a spectrum of the disease, rather than different clinical entities (BONNEMANN, 2011).
OBJECTIVES
This study aimed to describe the functionality of an individual with Bethlem Myopathy and one with Ullrich Congenital Muscular Dystrophy through specific tests, scales and skills assessment comparing these data with findings from the literature.
METHODS
Two retrospectives case studies were carried out in the period from 2009 to 2015. The evaluations were carried out at the Center for Human Genome Studies /USP, and the Project was submitted to the Ethics and Research Committee, numbered as 50667615.1.0000.5505.
There was minimal risk of the patient suffering a fall or any injury in the data collection room, even though they were accompanied by a researcher who took care that this did not happen.
No procedure was performed that would bring discomfort to the patient. In this study, an individual with MB and another DMCU carrier was evaluated at the Center for Human Genome Studies/USP.
The evaluation measures are muscle strength test, goniometry, lifting off the floor, chair lift, stair climbing, and gait. Four evaluations of each individual were used corresponding to the annual follow up of the same ones. A single duly trained evaluator performed the evaluation.
The strength test consisted of applying the Medical Research Council (MRC Index) that works by evaluating the level of activation performed by the muscle tested, being scored from 0 to 5. If the result obtained is no movement, the score will be 0; if there is a sketch, it will be 1, if there is a movement without gravity action, score 2, move against gravity 3, overcoming a slight resistance, score 4 and winning a maximum resistance, 5.
Goniometry is a way of assessing the articular angles of individuals, to identify the presence of retractions, contractures, and deformities.
The contractures were evaluated through this instrument and quantified according to their limitation.
The floor lift test consists of the individual sitting on a mat and getting up, being able to use compensations and supports if necessary. The lifting process is timed and time is the quantitative data used.
The chairlift test is about the individual starting from the seat in the chair of the sector for orthostatism, being able to make compensations and supports if need be. The task is timed to use this data as comparative for later evaluations.
Climbing stairs is an assessment of the time it takes the individual to climb the four-step path of a therapeutic ladder and can make compensations.
The gait test is a way of assessing the length of time the individual takes to walk a 10-meter hall.
The Vignos Scale and Egen Klassifikation were applied, if necessary.
The tests were applied by a single evaluator throughout the studied and adequately trained period.
It was performed through medical records analysis and direct interview.
The Vignos scale is a grading system based on functional capacity that serves to classify patients with muscular dystrophy. It is less accurate than the strength test, but it is easier to use and probably more effective for estimating muscle function.
Patients are graduated from 1 to 10, and the higher the rating, the higher the representation of increasing involvement. Being 1. when walking usually, with difficulty to run; 2. when there are detectable changes in posture or gait, climbing stairs without assistance of the handrail; 3. just upstairs with the handrail; 4. walks without external aid, does not climb stairs; 5. walks without external help, does not rise from the chair; 6. walks only with external assistance; 7. does not walk, sits upright in the chair without back, manages to drive the wheelchair, drinks and eats alone; 8. sit unsupported in the chair, can not drive it; 9. does not sit without wheelchair support, cannot eat or drink without assistance; 10. confined to bed, requires assistance for all activities (VIGNOS; SPENCER; ARCHIBALDI, 1963).
The Egen Klassifikation Scale is a scale developed in Denmark with the aim of quantifying the degree of functional limitation of patients with Duchenne muscular dystrophy with an advanced functional status. It is able to show the relationship between peripheral muscle strength, muscle contraction intensity, years of wheelchair use and forced vital capacity. Composed of 10 items, subdivided into 4 items, with scores ranging from 0 to 3, having these items as a way to evaluate from the ability to handle the wheelchair to the ability to remain in orthostatism and cough. It has a maximum total score of 30, and the higher the score, the worse the patient's condition. It was validated for Portuguese in 2006 by Martinez et al. (2006), at the Medical School of Ribeirão Preto at São Paulo.
RESULTS
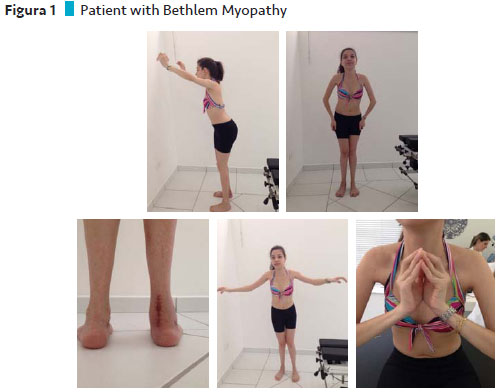
Case 1 description: Bethlem Myopathy
The patient with Bethlem's Myopathy is a young Caucasian, currently twenty years old, daughter of healthy parents, with normal neuromotor development for up to twelve months.
She presented the first symptoms at about one year of age, being frequent falls and difficulty getting up from the ground. Shortly afterward he began to walk on tiptoe, difficulty climbing stairs, running and jumping.
At the age of twelve, he began to have difficulty getting up and began to perform compensatory maneuvers for the activity, and without support, he already had a myopathic lift.
At the age of three, the investigations began, performed a muscular biopsy, electroneuromyography with decreased conduction velocity, indicating a myopathic condition, slightly increased CK (143). With these findings, some diagnostic possibilities were questioned, such as fascicular dystrophy, humeral scapular, and congenital myopathy.
At seventeen, he began to feel weaker, to lift with more difficulty, and to present joint contractures in feet, shoulders, and fingers, had an increase in the weakness of upper limbs and palpebral ptosis. He had hypotonia and hyperextensibility.
He performed four surgical procedures, the first in the calcaneal tendon bilaterally at seven years. The tendon on the left followed its growth correctly, but the right did not achieve the same result. He performed a new tenotomy on the calcaneus tendon and the right biceps femoris. At the age of 15, he restarted surgery on the tendon and at age 19, performed a tenotomy, an osteotomy, and a correction for the crooked right foot.
At eighteen, his lung function was evaluated. The findings indicated an FVC that oscillated between 45 and 50%. Since then, he has been following up at the Sono Institute's neuromuscular disease treatment clinic. It does not indicate Bipap at the moment, it does not have any symptoms and only performs stacking maneuver with ambu.
Some dermatological symptoms are present as follicular hyperkeratosis and tendency to form adherence in scars. The diagnosis was closed in 2014 with the NGS, obtaining a result with pathogenic alteration of c877G> A in heterozygous exon 10 of the COL6A1 gene, compatible with Bethlem Myopathy.
Its treatment is based on motor and water physical therapy twice a week, global postural re-education and acupuncture once a week. It uses supralodalic bracing and extensor splint bilaterally in other periods during the night. Currently, administered medications are alendronate, calcium, and vitamin D.
The muscular strength assessment was done through the MRC (Medical Research Council) and was analyzed in the four evaluations used for this study, with each individual.
The muscle groups that suffered the most losses during the four evaluations were flexors and extensors of the lower limbs, dorsiflexes and plantar flexors bilaterally, and in the first evaluation presented 90% of the MRC, in the second there was a decrease to 72.5%, maintaining scores on subsequent evaluations. In the second evaluation concerning the muscle groups corresponding to the flexors, extensors and shoulders abductors were obtained a score of 60%, in the following evaluations, there was a decrease to 53.3% of the MRC.
The muscle groups corresponding to flexors and elbow extensors, flexors and wrist extensors, obtained a slight decrease between the third and fourth evaluations, being 80% and 77.5% of the MRC, respectively. The only groups that remained unchanged were flexors, extensors and hip adductors with 60% of the MRC.
The bilateral (10º) hip flexion contractures were initially noted, which evolved to 20º, in knee flexion bilaterally (50º), progressing to -60º in the third evaluation and obtaining a return to 50º in the fourth evaluation. There was also equine contraction on the right, reaching 6º initially and evolving to 10º bilaterally in the third evaluation and the fourth, reaching to the right 14 and the left 10º. The elbow extension contracture was detected in the third evaluation and remained the same on the fourth (30º). New contractures were detected in the fourth evaluation in external rotation of the right shoulder (50º) and internal rotation of the hip bilaterally (10º).
All activities were timed and their values recorded. The floor lift test and the race evaluation were not testable since the first evaluation.
The chairlift had a worsening in time between the second and third evaluations being 6.84 "and 10.61" respectively and between the third and fourth evaluations there was an improvement in time from 10.61 "to 10.41".
The displacement by 10 meters showed a worsening from 17.90 "to 20.32" between the third and fourth evaluations.
The Vignos presented a score of 1 in all evaluations.
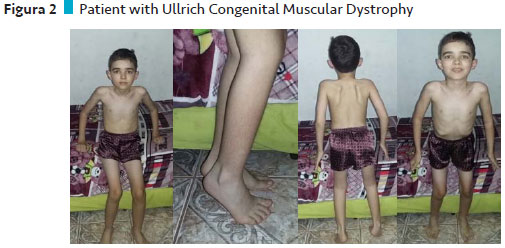
Case 2 description: Ullrich Muscular Congenital Dystrophy
Patient with Ullrich's Congenital Muscular Dystrophy, 12 years old, was born of normal birth, term gestation, without intercurrences. Parents are consanguineous cousins.
He presented a normal DNM, walked with 1 year and 2 months, but always suffered many falls and soon began to walk on tiptoe. He did not ride a bike, he did not run, he was rolling, he was climbing stairs, he had difficulty getting up off the ground. The speech was started early.
At 2 years of age, he consulted orthopedist who did not notice changes in the picture. After five years he was referred to a neurologist through another orthopedist.
At this time he only wandered with forefoot support, raised himself from the floor only with the aid of a chair with great difficulty, had diffuse amyotrophy in the lower limbs and proximal and distal upper limbs, hyperlordosis, ligament laxity and no contractures.
He was referred for DNA testing and muscle biopsy with the following diagnoses: Belly dystrophy, congenital muscular dystrophy, progressive spinal atrophy, and sarcoglycanopathy.
In the DNA test, two copies of exons 7 and 8 of the 5MN gene and two copies of 8 MN2 were found, ruling out the diagnosis of AEP.
In the immunohistochemical test in the muscular biopsy, the antibodies to dystrophin had a positive N-terminal region and C-terminal positive failed; antibodies to merosin and ɤ sarcoglycan exhibited usual labeling; the antibodies to α-sarcoglycan presented a positive. In Western Blot, the dystrophin band was present with the antibodies in the N-terminal and C-terminal regions.
Because of the partial deficiency of several proteins, it was difficult to differentiate the exact type of dystrophy. Still taking the probability of the diagnosis of muscular dystrophy of waists or congenital muscular dystrophy.
In 2014, NGS performed the diagnostic confirmation of a pathogenic mutation in homozygosis of exon 25 in the COL6A2 gene, and it was an autosomal dominant Ullrich congenital muscular dystrophy.
Its treatment is based on motor and aquatic physiotherapy once a week, occupational therapy also once a week and supraloral orthosis associated with extensor splint alternately, but it is difficult to adhere. Started the use of a wheelchair for about a year. Regarding the medications used caliamon kids and carvedilol.
Some muscle groups underwent more severe force reduction, with the most distal upper limbs, corresponding to flexors and elbow extensors, flexors and wrist extensors, in which they obtained 75% of the MRC in the first evaluation, in the second 70% in the third 65% and in the fourth 62.5%.
The proximal muscle groups of the lower limbs (flexors, extensors and hip adductors) also suffered a significant decline, with 56.7% in the first evaluation, 53.3% in the second, 50% in the third and 46.7% on Wednesday.
The proximal groups of the upper limbs (flexors, extensors, and abductors) started with 53.3%, obtained an improvement in the second evaluation with 60%, a new fall in the third with 53.3% and a new decline in the fourth to 43, 3%.
The distal muscle groups of the lower limbs (knee flexors and extensors, dorsiflexion and plantar flexors) reached the score of 75% initially, evolved negatively to 65% in the second and maintained a plateau of 70% in the last two evaluations.
The most intense and most evolving contractures during the four evaluations were knee flexion being 40º to the right, 30º to the left, 50º to 40º, 60º to 50º, 68º to 68º, respectively.
In the first evaluation, the hip flexion contracture presented 10º bilaterally, in the second and third evaluations they obtained an increase to 20º and in the fourth evaluation there was an increase in the right lower limb to 24º. Bilateral equine contraction remained the same during the period of the four evaluations, scoring 10º.
In the last evaluation, new contractures appeared in the internal rotation of the shoulder, being 28º to the right and 20º to the left, elbow flexion to the right in 30º and in internal rotation of the hip to the right with 10º and to the left in 4º.
The floor lift test improved between the first and third evaluations, being 29.96 "and 17", respectively.
The four-step climb test got a slight improvement from 6 "to 5.95" between the second and third.
In the displacement of 10 meters, there was a worsening between the second and fourth evaluations, being 15.14 "and 16.14", respectively.
The race was not testable since the first evaluation. In the latter, all tests were not feasible.
During the first three evaluations, he maintained a score of 2 at Vignos, and in the last one, there was a significant worsening in which he scored 7.
The Egen Klassifikation was used only in the last evaluation because the individual became a wheelchair, obtaining a score of 3.
DISCUSSION
The diseases Bethlem Myopathy and Congenital Muscular Dystrophy of Ullrich are poorly described in the literature from the functional point of view, being the majority related to genetic and descriptive questions only. It is believed that this happens because they are rare and have a difficult diagnose.
In the case of Bethlem myopathy, a less significant loss was observed, and the groups most affected were those belonging to the scapular cingulate and lower limbs, which is confirmed by Bonnemann (2011), treating MB as a pathology with milder intensity of symptoms, even though a form of collagen type VI myopathy. Another study by Deconinck et al. confirms the findings, in which 36 MB patients were evaluated in the following categories: muscular strength, disease evolution, respiratory evolution, muscle biopsy, COLVI secretion in fibroblasts culture and genotype-phenotype correlation. It was observed that 31 patients had muscle strength deficit, concluding that the groups that were most affected were the scapular cingulate, the lower limbs, and practically all distally in the feet and hands (DECONINK et al., 2015).
In the case of Ullrich's Congenital Muscular Dystrophy, a significant decrease of the generalized muscular force was observed, with emphasis on scapular cingulate, upper limbs and pelvic cingulate. Similar to the data concluded in these assessments, a study by Miscione et al. (2013) evaluated the body composition, muscular strength, and functionality of 7 MB and 1 MBM patients. In order to evaluate muscle strength, the make test was performed, and it was concluded that in these individuals it was reduced in all groups when compared to normal values, and it was that the most affected were knee extensors and elbow flexors, but were not separated according to their pathologies being generated a redundant statistic for all individuals evaluated (MISCIONE et al., 2013).
In these two cases, there was a reduction in generalized muscle strength, with the scapular cingulate being commonly affected. However, what happens when comparing these two individuals is that the progression of these deficits goes differently, in which Bethlem's Myopathy takes a slower slope, and Ullrich's Congenital Muscular Dystrophy follows drastically more severe.
Thus, both have significant losses of muscle strength, and disease staging may be closely related to these findings, which, despite having different degrees of impairment in their complete course, are sharpened about the presented symptomatology.
In the case of Bethlem myopathy, there was an asymmetrical contracture, with a greater right involvement, appearing in the plantar flexors and the external rotation of the shoulder. Another segment that remains in evolution is the hip flexion and remains constant in elbow extensors.
To confirm these findings, another case of study of Park et al. (2015) described the characteristics of a 38-year-old woman with a diagnosis of Bethlem Myopathy, with the main picture of proximal weakness and ankle contractures being exposed. Corroborating with this study, Allamand et al. (2011) cites the main segments affected in his review as the flexors of elbows, wrists and fingers, plus plantar flexors.
No studies were found describing proximal contractures as observed in the evaluated individual.
In the case of Ullrich's Congenital Muscular Dystrophy, several more severe contractures were found in knee flexion with an evolutionary character, maintains a constant equine, and has an asymmetric characteristic with greater right compromise in hip flexion, internal rotation of the shoulder, flexion of the elbow and internal hip rotation.
In keeping with the same line and agreeing with the findings of the patient of our study, a study by Park et al. (2014) cites that the segments that suffer more contractures are hips, leading to an increase of flexion and generating an equine pattern in the ankles. In another study by Demir et al. (2002), five patients and 11 individuals from 3 consanguineous families of patients were investigated. They were evaluated from the genetic and phenotypic point of view, with lower limbs tending to be more affected and in most cases, hip flexor contractures are installed. They also concluded that between 8 and 12 years of age, the frames of proximal contractions of lower limbs and in equine intensify. One of the patients required a surgical procedure for equine correction. Retractions in toe flexion were also observed in another patient (DEMIR et al., 2002).
The two cases were characterized by severe contractures in several segments, and in both cases, the severity was higher in knee flexion, and the asymmetrical character was also notorious, in which the right hemisphere was more affected. In contrast, the progression of severity was much more intense in the individual with DMCU, added too much higher variability of sites.
In this way, contractures are common causes of inactivity and functional losses in the two diseases, being the percussion of other musculoskeletal and respiratory changes.
In Bethlem myopathy, there was a progressive worsening of motor skills, with the ground-lift test and the race never performed, and the tests of getting up from the chair, climbing stairs and walking for 10 meters had their times relatively increased. This information is based on a study by Foley et al. (2009), which described two cases with MB. The first was a 43-year-old patient with proximal weakness and contractures, but in gait evaluation, he was able to walk without an auxiliary device, only performing compensations in Trendelemburg.
The other individual was a biological similarity of the first and had the same motor pattern (FOLEY et al., 2009). In the case of Ullrich's Congenital Muscular Dystrophy, there was a much more aggressive worsening, and his walking pattern was never evaluated, and in the last evaluation, he was not able to perform any activity. In a study by Zhang et al. (2014), eight patients with DMCU were evaluated, with 5 of these individuals acquiring gait but had significant motor difficulties and three never acquired this motor frame, illustrating the severity of this disease and correlating with subsequent motor losses (ZHANG et al., 2014).
In both cases, motor functional losses occur, with Bethem's myopathy being milder in relation to Ullrich's Congenital Muscular Dystrophy. The loss of gait in the second case is much more precocious caused by the loss of more incisive muscle strength and more severe contractures.
CONCLUSION
Significant losses of muscle strength were observed along the trajectory of these individuals, evidencing the need to emphasize the follow-up of these evolutions in order to observe the fugacity of the presented deficits and to generate better strategies for treatment.
The findings of the literature coincide with what was evaluated in the individuals, with contractures in common between the patients of both DMCU and MB, and each individual can be more or less affected according to the natural evolution but can be minimized with proper care.
There is still a need for further studies relating MB and DMCU with specific motor evaluations, such as those studied in these two cases. It is concluded that the evaluation of skills for the targeting of the treatment and correlating with the variability of motor losses in each individual is extremely important.
REFERENCES
ALLAMAND, V. et al. ColVi myopathies: where do we stand, where do we go? Skeletal muscle, v. 1, n. 30, p. 1-13, Sept. 2011. [ Links ]
BETHLEM, J.; WIJNGAARDEN, G. K. Benign myopathy, with autosomal dominant inheritance. A report on three pedigrees. Brain, v. 99, n. 1, p. 91-100, Mar. 1976. [ Links ]
BONNEMANN, C. The collagen VI-related myopathies: muscle meets its matrix. Nature Reviews, v. 7, n. 7, p. 379-390, June 2011. [ Links ]
CARAKUSHANSKY, G.; RIBEIRO, M. G.; KAHN, E. Moderately progressive Ullrich congenital muscular dystrophy. Jornal de Pediatria, v. 88, n. 1, p. 93-96, Jan./Feb. 2012. doi: http://dx.doi.org/10.2223/JPED.2112 [ Links ]
DECONINK, N. et al. Bethlem Myopathy: long-term follow up identifies Col6 mutations predicting severe clinical evolution. J. Neurol. Neurosurg. Psychiatry, v. 86, n. 12, p. 1337-1346, Dec. 2015. [ Links ]
DEMIR, E et al. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich Congenital Muscular Dystrophy. Am. J. Hum. Genet, v. 70, n. 6, p. 1446-1458, June 2002. doi: https://doi.org/10.1086/340608 [ Links ]
FOLEY, A. R. et al. Autosomal recessive inheritance of classic Bethlem Myopathy. Neuromuscul. Disord., v. 19, n. 12, p. 813-817, Dec. 2009. doi: 10.1016/j.nmd.2009.09.010 [ Links ]
MARTINEZ, J. A. B. et al. Validação da escala motora EK para a língua portuguesa. Rev. Assoc. Med. Bras., São Paulo, v. 52, n. 5, p. 347-351, set./out. 2006. doi: http://dx.doi.org/10.1590/S0104-42302006000500024 [ Links ]
MISCIONE, M. T. et al. Body composition, muscle strength, and physical function of patients with Bethlem Myopathy and Ullrich Congenital Muscular Dystrophy. The Scientific World Journal, v. 2013, p. 1-6, 2013. doi: http://dx.doi.org/10.1155/2013/152684 [ Links ]
PALMA, S. et al. Muscle proteomics reveals novel insights into the pathophysiological mechanisms of collagen VI myopathies. Journal of Proteome Research, v. 13, n. 11, p. 5022-5030, Sept. 2014. doi: 10.1021/pr500675e [ Links ]
PARK, Y. et al. Ullrich Congenital Muscular Dystrophy possibly related with COL6A1 p.Gly302Arg variant. Annals of Reabilitativo Medicine, v. 38, n. 2, p. 292-296, Apr. 2014. doi: 10.5535/arm.2014.38.2.292 [ Links ]
PARK, H. J. et al. Molecular genetic diagnosis of a Bethlem Myopathy Family with an autosomal dominant COL6A1 mutation, as evidenced by exome sequencing. J. Clin. Neurol., v. 11, n. 2, p. 183-187, Apr. 2015. doi: 10.3988/jcn.2015.11.2.183 [ Links ]
REED, U. C. et al. Ullrich Congenital Muscular Dystrophy and Bethlem Myopathy: Clinical and genetic heterogeneity. Arq. Neuropsiquiatr., São Paulo, v. 63, n. 3, p. 785-790, Sept. 2005 doi: http://dx.doi.org/10.1590/S0004-282X2005000500013 [ Links ]
REED, U. C. Congenital Muscular Dystrophy. Part I: a review of phenotypical and diagnostic aspects. Arq. Neuropsiquiatr., v. 67, n. 1, p. 144-168, Mar. 2009. doi: http://dx.doi.org/10.1590/S0004-282X2009000100038 [ Links ]
ROZORGMEHR, B. et al. Ullrich Congenital Muscular Dystrophy (UMCD): clinical and genetic correlations. Iran J. Child. Neurol., v. 7, n. 3, p. 15-22, Summer 2013. [ Links ]
TAGLIAVINI, F. et al. Defective collagen VI alfa 6 chain expression in the skeletal muscle of patients with collagen VI-related myopathies. Biochimica et Biophysica Acta, v. 1842, n. 9, p. 1604-1613, Sept. 2014. doi: 10.1016/j.bbadis.2014.05.033 [ Links ]
TONI, S. et al. Nutritional status evaluation in patients affected by Bethlem Myopathy and Ullrich Congenital Muscular Dystrophy. Frontiers in Aging Neuroscience, v. 6, n. 315, Nov. 2014. doi: 10.3389/fnagi.2014.00315 [ Links ]
VIGNOS, P. J.; SPENCER, G. E.; ARCHIBALDI, K. C. Management of progressive muscular dystrophy of childhood. JAMA, v. 184, n. 2, p. 89-96, 1963. [ Links ]
VOERMANS, N. C. et al. Joint hypermobility as a distinctive feature in the differential diagnosis of myopathies. J. Neurol., v. 256, n. 1, p. 13-27, 2009. [ Links ]
ZAMURS, L. K. et al. Aberrant mitochondria in Bethlem Myopathy patient with a homozygous amino acid substitution that destabilizes the collagen VI alfa 2 (VI) chain. Journal of Biological Chemistry, v. 290, n. 7, p. 4272-7251, 2015. [ Links ]
YONEKAWA, T.; NISHINO, I. Ullrich Congenital Muscular Dystrophy: Clinicopathological features, natural history and pathomechanisms. J. Neurol. Neurosurg. Psychiatry, v. 86, n. 3, p. 280-287, Mar. 2015. [ Links ]
ZHANG, Y. Z. et al. Novel collagen VI mutations identified in Chinese patients with congenital muscular dystrophy. World J. Pediatr., v. 10, n. 2, p. 126-132, May 2014. [ Links ]
ZOU, Y. et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Human Molecular Genetics, v. 23, n. 9, p. 2339-2352, May 2014. doi: 10.1093/hmg/ddt627 [ Links ]
Recebido em: 10.11.2017
Aprovado em: 30.11.2017