









Serviços Personalizados
Journal

artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores
Compartilhar

 Permalink
PermalinkJournal of Human Growth and Development
versão impressa ISSN 0104-1282versão On-line ISSN 2175-3598
J. Hum. Growth Dev. vol.30 no.3 São Paulo set./dez. 2020
https://doi.org/10.7322/jhgd.v30.11118
ORIGINAL ARTICLE
The numerous denominations of the Brugada syndrome and proposal about how to put an end to an old controversy - a historical-critical perspective
Joseane Elza Tonussi MendesI; Kjell NikusII; Raimundo Barbosa-BarrosIII; Andrés Ricardo Pérez-RieraI
ILaboratorio de Delineamento de Estudos e Escrita Científica, Centro Universitário Saúde ABC, Santo André, São Paulo, Brazil
IIHeart Center, Tampere University Hospital and Faculty of Medicine and Health Technology, Tampere University, Finland
IIICoronary Center of the Hospital de Messejana Dr. Carlos Alberto Studart Gomes, Fortaleza, Ceará, Brazil
ABSTRACT
BACKGROUNG: The eponymous Brugada Syndrome (BrS) in honor of its discovery as an independent entity by the Spanish/Catalan Brugada brothers, Pedro and Josep, has deserved numerous denominations derived mainly from the clinical genotype/phenotype correlation. The purpose of this manuscript is to present and analyze the nomenclatures that this intriguing and challenging syndrome has received over the past 28 years. We also compared the main features between cases from the first report of the Brugada brothers and an article by Martini et al. The nomenclatures used by these authors are closely linked to the BrS, but the cases (except one) presented in the article by Martini et al do not present the type 1 Brugada ECG pattern, which is mandatory for the diagnosis of BrS
Keywords: Brugada syndrome, eponymous, nomenclature, electrocardiographic hallmark.
RESUMO
INTRODUÇÃO: A Síndrome de Brugada (SB), em homenagem à sua descoberta como entidade independente pelos irmãos espanhóis / catalães Pedro e Josep Brugada, tem merecido inúmeras denominações derivadas principalmente da correlação genótipo /fenótipo clínico. O objetivo deste manuscrito é apresentar e analisar as nomenclaturas que esta intrigante e desafiadora síndrome recebeu nos últimos 28 anos. Também comparamos as principais características entre os casos do primeiro relato dos irmãos Brugada e um artigo de Martini e col. As nomenclaturas utilizadas por esses autores estão intimamente ligadas à SB, mas os casos (exceto um) apresentados no artigo de Martini e cols. não apresentam o padrão eletrocardiográfico de Brugada tipo 1, obrigatório para o diagnóstico da SB
Palavras-chave: Brugada syndrome, eponymous, nomenclature, electrocardiographic hallmark.
Authors summary
Why was this study done?
The main reason is that there is still a dispute over the "paternity" of Brugada syndrome, consequently we made a comparative analysis of the disputed manuscripts. We also highlight the numerous names of Brugada syndrome, since many colleagues ignore most of those names.
What did the researchers do and find?
We confirm that Brugada syndrome is the same entity as sudden night death, highly prevalent in Southeast Asian countries (Thailand, Philippines and Japan), both of which share mutations in the same gene: SCN5A. In addition, Asian patients have an identical electrocardiographic pattern.
What do these findings mean?
These discoveries allow us to affirm with tranquility that the Brugada brothers were the true discoverers of the syndrome, considering the claim of the Italian authors inadequate. Even so, the dispute considering Brugada syndrome as an independent entity or forming part of the spectrum of arrhythmogenic right ventricular cardiomyopathy is still controversial.
INTRODUCTION
The last "new clinical-cardiologic syndrome" described in the 20th century, named as the eponymous Brugada Syndrome (BrS) in honor of its discovery as an independent entity by the Spanish/Catalan Brugada brothers, Pedro and Josep, has deserved numerous denominations derived mainly from the clinical genotype/phenotype correlation. The purpose of this manuscript is to present and analyze the nomenclatures that this intriguing and challenging syndrome has received over the past 28 years. Interestingly, this entity had been described for the first time more than 100 years ago (1917).
Below is a list of the main nomenclatures found in the literature to describe BrS.
• Brugada syndrome: In 1986, Prof. Pedro Brugada-Terradelas had a Caucasian patient of Polish origin, a child with distinct electrocardiographic (ECG) findings, who suffered from repetitive syncopal episodes. The family background of the young boy revealed that a sister had suffered sudden cardiac death (SCD), even though she had been treated with pacemaker implantation and amiodarone. In 1991, Pedro and Josep Brugada, adding two more cases, presented an abstract in the North American Society of Pacing and Electrophysiology (NASPE) meeting, describing their findnigs as a "new clinical-cardiologic syndrome''. In 1992, they decribed the ECG findings as an association of right bundle branch block (RBBB), persistent ST segment elevation, normal QT interval and SCD1. The series consisted of eight Caucasian patients (six male and two female), six adults and two children, with recurrent episodes of recovered SCD. Clinical data, findings in the ECGs, 24-hour Holter monitoring, thallium stress test, transthoracic echocardiography, chest roentgenograms, laboratory data, coronary angiography with right and left ventriculography, electrophysiological study (EPS), programmed ventricular stimulation and endomyocardial biopsy were available in most cases with a follow-up of five years.
The ECG of the patients showed normal PR intervals, RBBB pattern, persistent ST segment elevation in the right precordial leads V1 to V2-V3 not explained by electrolyte disturbances, ischemia or structural heart disease and normal QT interval. No histologic abnormalities were found on biopsy performed in four patients. The arrhythmia leading to (aborted) SCD was a fast and polymorphic ventricular tachycardia (PVT) always initiating after a short, coupled premature ventricular contraction (PVC). A similar arrhythmia was initiated by two to three ventricular extrastimuli in four of the seven patients studied by programmed electrical stimulation. Four patients had a prolonged HV interval. Four patients received an implantable cardioverter defibrillator (ICD), all of them alive at the end of follow-up; in one of the patients, ICD activity was recorded during follow-up. Common clinical and ECG features defined a distinct idiopathic syndrome in this group of patients. The eponym "Brugada syndrome" was coined and documented for the first time in January 1996 by Yan and Antzelevitch2 and during the same year also by Kobayashi et al3.
The first mutation in the SCN5A gene associated with the syndrome, was identifed in 19984, two years after the name of the eponym by Yang and Antezelevich.
• Lai-Tai (in Thailand and Laos)5, meaning "death during sleep". Sudden unexplained death syndrome (SUDS)6 was noted in 1977 among southeast Asian Hmong refugees in the United States and Canada7,8.Between 1981 and 1988, the Centers for Disease Control and Prevention reported a very high incidence of sudden death among young male Southeast Asians who died unexpectedly during sleep9. The pattern of death has long been prevalent in Southeast Asia. The disease was noted in Singapore, when a retrospective survey of records showed that 230 otherwise healthy Thai foreign workers residing in Singapore died suddenly of unexplained causes between 1982 and 1990. The victims (all males) were aged 21-54 (median 34). 80% came from villages in North-eastern Thailand. The median interval between arrival in Singapore and death was eigth months. All the victims were apparently healthy before going to bed. When deaths were witnessed, room-mates were alerted or awakened by noisy breathing, gasping, or groaning, but bystander resuscitation attempts were unsuccesful. In some cases the body was rigid and fists clenched, and there was urinary incontinence10. In 1997 Nadenamanee et al. studied 27 young adult Thai men (mean age, 39.7±11 years) referred for cardiac arrest due to ventricular fibrillation (VF), usually occurring at night while asleep (n=17), or for symptoms similar to the clinical presentation of SUDS (n=10). They performed EPS and cardiac catheterization. Of the patients without structural heart disease (except 3 patients with mild left ventricular hypertrophy), 16 had a distinct ECG abnormality consisting of RBBB and ST segment elevation in the right precordial leads, while 11 patients had a normal ECG11. Table 1 shows the distinct clinical differences, including the risk for SUDS, between the two groups.
In 2015, Nadenamanee et al. also studied six whole hearts from young male post-mortem cases of SUDS (mean age 23.2 years) and familial BrS with negative cardiac autopsy and matched the hearts to six homograft control hearts by sex and age (within 3 years) and by random risk set sampling. Cardiac autopsy sections from cases and control hearts were stained with picrosirius red for collagen. The right ventricular outflow tract (RVOT) was studied in detail, including immunofluorescent stain for connexin-43 (Cx43). Collagen and Cx43 were quantified and compared. An in vivo study was undertaken on six consecutive BrS patients (mean age 39.8 years, all men) during epicardial RVOT ablation via thoracotomy. Abnormal late potentials and fractionated potentials indicative of dromotropic disturbances were identified, and biopsies were taken before ablation. Collagen was increased in the BrS autopsy cases compared with control hearts. Fibrosis was most evident in the RVOT and the epicardium. Cx43 expression was reduced in the RVOT of the BrS hearts. Autopsy and in vivo RVOT samples identified epicardial and interstitial fibrosis. This was collocated with abnormal potentials in vivo, and after ablation the type 1 Brugada ECG pattern was abolished, and no ventricular arrhythmias were detected during 24.6±9.7 month follow-up. The authors concluded that BrS is associated with epicardial surface and interstitial fibrosis and reduced Cx43 expression in the RVOT. This collocates to abnormal potentials, and their ablation abolishes the type 1 Brugada ECG pattern and life-threatening arrhythmias. Additionally, this entity is associated with increased collagen in all cardiac chambers12. Based on these findings, BrS should be considered as a cardiomyopathy and not as an electrical heart disease.
• Nightmares bangungut (Philippines). Bangungot is known by many medical names: sudden arrhythmic death syndrome or sudden arrhythmia death syndrome (SADS), bed death, SUDS, BrS, and sudden unexpected nocturnal death syndrome (SUNDS)13. Vatta et al. in 2002 demonstrated for the first time that SUNDS and BrS are allelic entities14. The authors considered Bangungut/SUNDS and BrS as the same disorder phenotypically, functionally, and genetically, as has been asserted for cases of Lai-Tai in Thailand and Pokkuri in Japan.
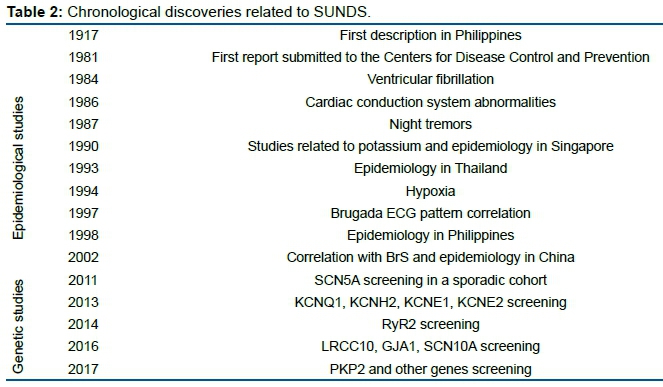
Bangungot is the Tagalog word for "nightmare," but it can also refer to a malevolent spirit elemental in Filipino folklore. Also known in Ilocano (a Philippine ethnolinguistic group) as batibat, it appears in the shape of an overweight man (bangungot) or overweight Botero-like woman (batibat) who sits on the chest or head of the victim (always male) and suffocates him to death. According to witnesses, the victims groan (ungol in Tagalog), while struggling against the bangungot, and if they do not rise from sleep, (bangon in Tagalog), they die. It is useless to try to push away the bangungot or batibat, but one can survive if one is able to wiggle a big toe and wake up. A Foundation for Bangungot Research was set up in the 1950s by the owner of Benipayo Press after his son died of bangungot. Table 2 Lists the chronological discoveries of diseases related to SUNDS.
It is more than a century since the first description of SUNDS in Philippines in 191715. There were two major active periods of studies on SUNDS. The early stage mainly focused on collecting the epidemiological characteristics, and revealing the probable external environmental risk factors, while the later stage aimed at uncovering the intrinsic gene susceptibility.
• Pokkuri death syndrome (PDS) is a SUNDS in Japan. Okada et al., in order to clarify the importance of the conduction system involvement in cases of SCD, preformed autopsies in 35 hearts obtained from patients, who died within one hour after the onset of symptoms. The hearts were examined histopathologically and compared with 27 age- and disease-matched and 30 only age-matched control hearts from individuals, who had not died suddenly. The conduction system was serially sectioned using Lev's method and observed under a light microscope. Purkinje cell lesions, which made a structural maze compatible to the electrophysiological re-entry mechanism, abnormalities of the atrioventricular (AV) node artery and hypertrophy of the AV node and the bundle of His were more prominent in the SCD group with long QT syndrome, idiopathic VF and AV and bundle branch blocks due to both ischemic heart and myocardial diseases. Sinus node fibrosis with minor anomalies of the sinus node artery was specifically seen in the SCD of PDS. The same lesion, but with sclerosis of the sinus node artery, was seen in the cases of SCD with hypertensive heart disease16. Most of the PDS cases autopsied in Japan have been found among young males without any specific known disease, who died suddenly around midnight with convulsions. The underlying cause of PDS may be a genetic disorder(s) that is more prevalent in South Asian populations. The relationship between the narrow circumferences of coronary arteries without atherosclerosis and hyperlipoproteinemia associated with the risk of coronary artery spasm needs to be further investigated in a larger number of PDS cases17.
• Idiopathic VF of the Brugada type. The clinical characteristics of the Brugada type idiopathic VF (right ventricular abnormalities with lipid accumulation, familial occurrence, male predominance) resembles those of arrhythmogenic cardiomyopathy (ACM)18,19, but there are differences between the two diseases. ST elevation in the precordial leads is one of the characteristics in the Brugada type idiopathic VF, while this characteristic is rarely found in patients with typical ACM. HV prolongation or RBBB is a common finding in patients with Brugada type idiopathic VF3, and conduction abnormalities are usually confined to the right ventricular myocardium, manifested by delayed potential and fragmentation20.
• SUDS. BrS is currently considered a genetic/environmental condition that causes severe disturbances in cardiac rhythm and may result in SUDS in an apparently healthy person. The mild structural changes in the heart are typically located in the RVOT although the disease may affect the entire heart12,21,22. BrS is a major cause of SUDS, also known as SADS. Dhamapurkar et al. described a case of a 25-year-old man diagnosed with BrS following a nose bleed, who had non-fatal cardiac arrest23. He was left with a visual impairment, dystonia, hypersensitivity, and language and cognitive dysfunction. The authors pointed out the importance of comprehensive patient care, including work on family basis, focus on meaningful functional activity, finding the right compensatory strategy that can help overcome communication, cognitive or emotional impairments, providing therapy within a safe therapeutic milieu, providing appropriate models of psychological therapy, and effective approach to sharing our understanding of the client within the interdisciplinary team and the wider system.
• SUNDS: Huang et al. identified p.E129K as a potential pathogenic mutation underlying the death of one patient with SUNDS. The authors also provided evidence to show that a rare variant in East Asia (p.P69L) might contribute to the genetic cause for one SUNDS victim and two family members with BrS. This was the first report of genetic screening of LRRC10 in Chinese SUNDS and BrS. The investigation implies that LRRC10 may be a new susceptible gene for SUNDS with genetical links of LRRC10 variants to primary arrhythmia-associated BrS and SUNDS. The authors planned to perform EPS studies to test the functional impact of these putative pathogenic mutations of LRRC10 to find data to support their role as pathogenic variants in Chinese SUNDS and BrS. Identifying p.E129K might be a potential pathogenic mutation underlying SUNDS24.
LRRC10 is a protein-coding gene. Diseases associated with LRRC10 include dilated cardiomyopathy and SUNDS. Annotations related to this gene include actin binding and alpha-actinin binding. An important paralog (a gene that is related to another gene in the same organism by descending from a single ancestral gene that was duplicated and that may have a different DNA sequence and biological function) of this gene is LRRC108.
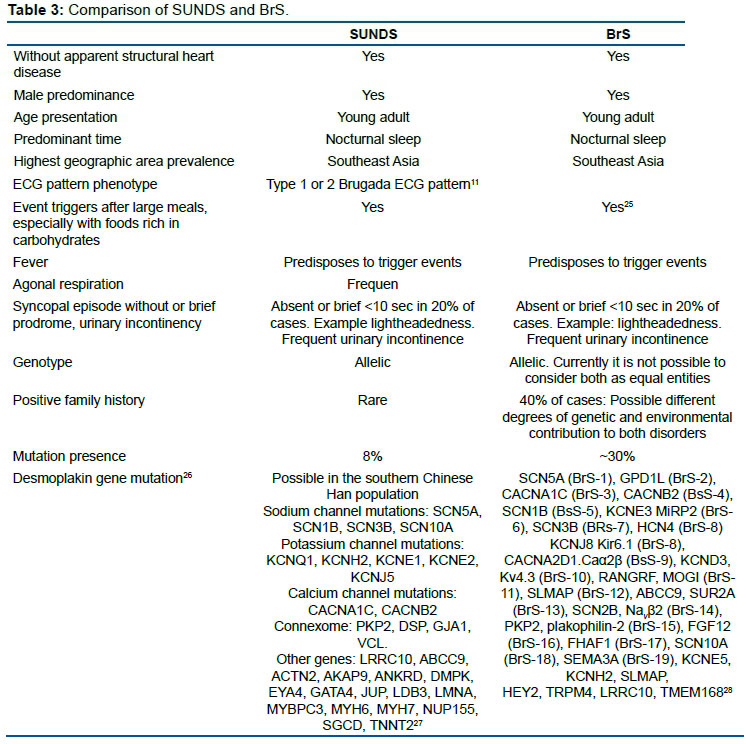
Is SUNDS identical to BrS? Table 3 compares both entities.
Important note: Currently the only gene considered as causally related to BrS is SCN5A. Except for SCN5A, rare coding variation in previously reported arrhythmia-susceptibility genes did not contribute significantly to the occurrence of BrS in a population with European ancestry.
Extreme caution should thus be taken when interpreting genetic variation in molecular diagnostic setting, since rare coding variants were observed in a similar extent among cases versus controls, for most previously reported BrS-susceptibility genes29. Genetic testing for the BrS is controversial since ~ 20% of patients can be identified with the causing gene mutations, most of which are in SCN5A.
Furthermore, even in the SCN5A mutation-positive carriers, their phenotypes are not completely consistent with BrS, but may cause other inherited arrhythmogenic disease including BrS, LQT3, cardiac conduction defect, sick sinus syndrome, atrial fibrillation, dilated cardiomyopathy. progressive cardiac conduction defect, multifocal ectopic Purkinje-related premature contraction, and the onset of a variety of non-cardiac diseases, including irritable bowel syndrome30, myotonic dystrophy31, epilepsy32, pain33, and ataxia34. On the other hand, a recent Japanese BrS registry demonstrated that the pore-region mutations in SCN5A are significantly associated with a risk of lethal cardiac events. Furthermore, a genome-wide association study revealed that a common variant in SCN10A or HEY2 in addition to SCN5A is associated with BrS.
Consequently, BrS may not be a monogenic Mendelian disease but an oligogenic channelopathy35. Genetic testing should never be used to make a primary diagnosis of BrS in isolation, especially when the clinical phenotype of BrS is absent.
While guidelines have provided a major effort towards the standardization of variant interpretation, inter-laboratory differences in the use and implementation of the American College of Medical Genetics and Genomics guidelines criteria exist. Some genetic testing companies still conflate the mere presence of a specific variant in an early paper as evidence for pathogenicity, despite these reports lacking adequate numbers of healthy controls, functional characterization of the variant, or illustration of proper co-segregation with the disease phenotype within a multigenerational pedigree; each of which individually, and more so concomitantly, constitutes more definitive evidence in the interrogation of a potentially pathogenic variant.
Campuzano et al. aimed to clarify the role of all currently reported variants in minor genes associated with BrS. They performed a comprehensive analysis according to the American College of Medical Genetics and Genomics guidelines of published clinical and basic data on all genes (other than SCN5A) related to BrS.
Their results identified 133 rare variants potentially associated with BrS. After applying current recommendations, only six variants showed a conclusive pathogenic role. All definitively pathogenic variants were located in four genes encoding sodium channels or related proteins SCN5A, SLMAP, SEMA3A, SCNN1A, and SCN2B for BrS36.
• Brugi-Brugi syndrome. This term was introduced during a cardiology conference on SCD organized by the Brugada brothers in July 1999, held in the Cardiovascular Center OLV Hospital in Aalst, Belgium37.
From our point of view, it is not ideal to name medical entities using the eponyms of their discoverers because this practice does not contribute to the understanding of the disease. For example, the long QT syndrome is no longer Roman-Ward syndrome, and early repolarization syndrome is not Haissaguerre syndrome.
Literature polemic between Brugada syndrome versus Nava-Martini-Thiene syndrome
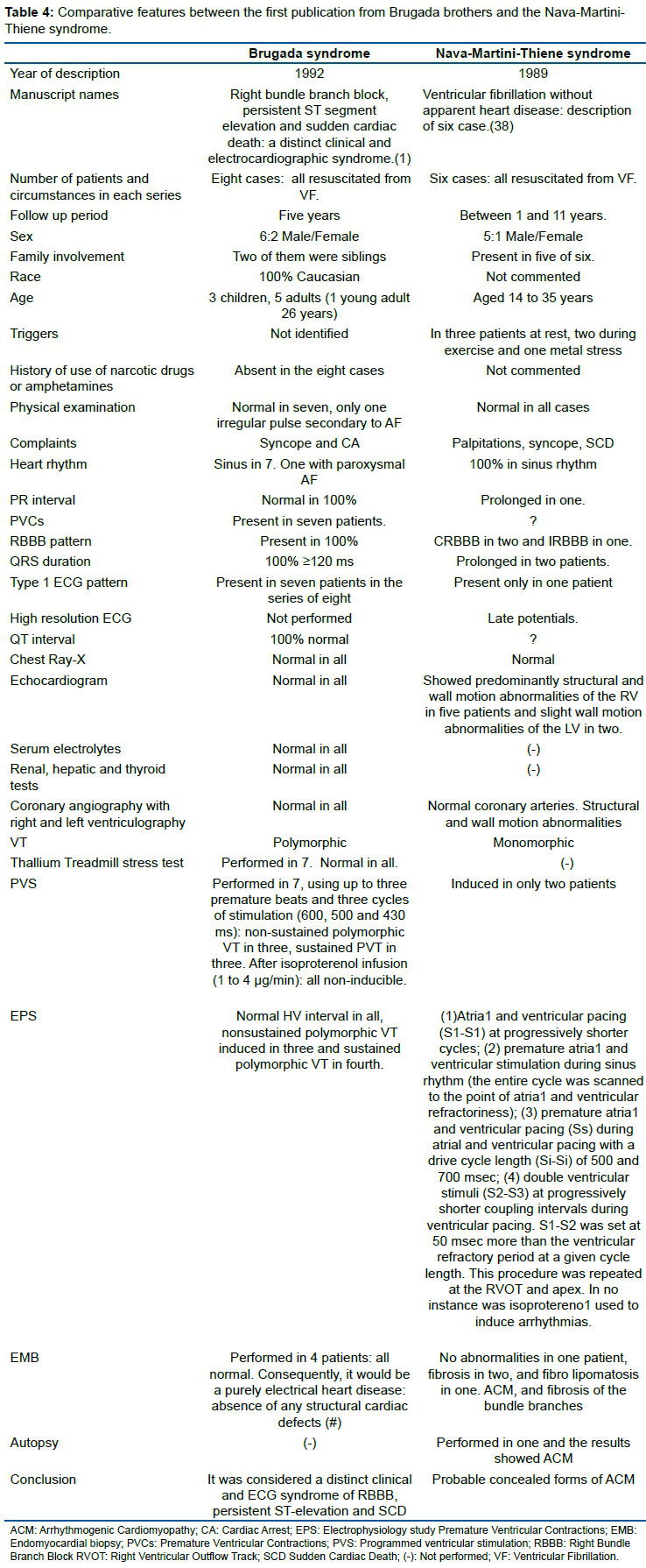
There has been debate in the literature regarding similarities between the BrS and a syndrome of RBBB, ST elevation and sudden death described by Italian doctors. Table 4 shows comparative features between the first publication from Brugada brothers and the Nava-Martini-Thiene syndrome.
(#) The Brugada brothers presented the entity as a pure electrical condition (and keep doing that to discriminate it from ACM) but nowadays there is no doubt that BrS is in the spectrum of ACM. The spectrum of the ACM is wider than initially thought and usually referred to with the adjective "right ventricular", with the evidence of biventricular or even isolated left ventricular forms, so that it is increasingly identified simply as "arrhythmogenic cardiomyopathy". There is not a single heart of a true BrS which is completely normal in the published reports. The entity is an electrical disease with minor structural changes39. Consequently, we should suspect that the histological analysis performed on four of the eight hearts may not have been detected, because the structural changes are extremely subtle, or could have been performed at an early stage. If BrS is considered a part of the spectrum of ACM, BrS does not exist as an independent entity. It would just be a concealed form of ACM40. Consequently, at our discretion, it is a contradiction of the 2013 expert consensus statement on inherited primary arrhythmia syndromes, the Heart Rhythm Society/European Heart Rhythm Association/Asia Pacific Heart Rhythm Society (HRS/EHRA/APHRS), because in this consensus, BrS was considered an independent entity within the inherited arrhythmia syndromes.
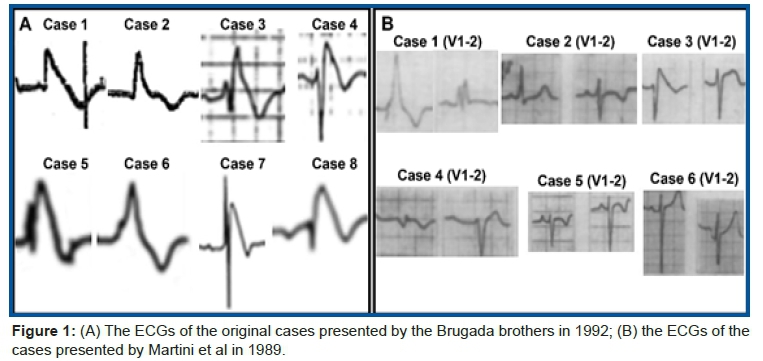
Figure 1 shows a comparison between Brugada and Martini ECGs in the right precordial leads.
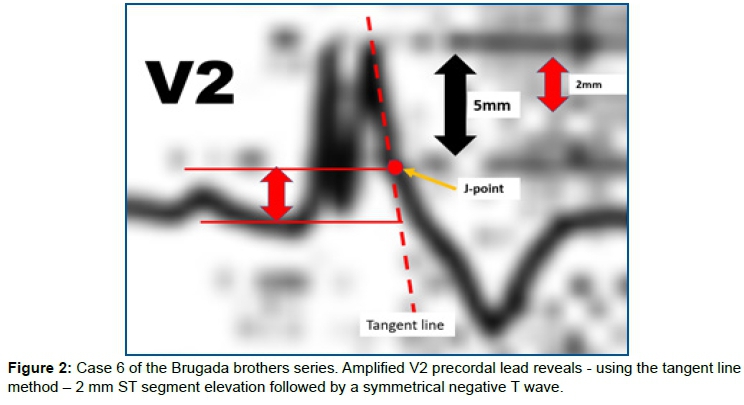
In the paper by the Brugada brothers, all patients, except case 2, showed spontaneous type 1 Brugada ECG pattern in the right precordial leads V1 and V2 with the ECG electrodes positioned at the 4th intercostal space. Additionally, the case number 6 of their series can be considered doubtful; however, when amplifying the precordial lead V2, we notice that it fulfills the criteria of the Type 1 Brugada ECG phenotype, because the ST segment elevation is exactly at 2 mm from the J point followed by a symmetrical negative T wave (Figure 2).
On the other hand, in the paper by Martini et al, only the ECG of patient case 3 shows the type 1 Brugada ECG pattern. Consequently, almost all ECGs in the small series of the Italian researchers do not fulfill the fundamental criterion for the diagnosis of BrS following the Expert Consensus Recommendations on BrS (2013)41:
1) BrS is diagnosed in patients with ST-segment elevation with type I morphology ≥2 mm in ≥1 lead among the right precordial leads V1 and V2 positioned in the 2nd, 3rd, or 4th intercostal space, occurring either spontaneously or after provocative drug test with intravenous administration of Class I antiarrhythmic drugs.
2) BrS is diagnosed in patients with type 2 or type 3 ST-segment elevation in ≥1 lead among the right precordial leads V1 and V2 positioned in the 2nd, 3rd, or 4th intercostal space when a provocative drug test with intravenous administration of Class I antiarrhythmic drugs induces a type I ECG morphology.
As the eight authors of Martini manuscript are from Padua, Italy, where the prevalence of ACM is high, it would not be surprising if the six studied patients turned out to have a concealed form of ACM, which is frequently confused with BrS because the type 1 Brugada ECG pattern can be seen in this variety of cardiomyopathy. Giovanni Maria Lancisi, in the book entitled De Motu Cordis et Aneurysmatibus, Caput V, De Hereditaria ad Cordis Aneurysmata Constitutione: De Cordis Prolapse (an hereditary predisposition to cardiac aneurysm and bulgings), described the history of a family with disease recurrence in four generations, presenting with palpitations, dilatation and aneurysms of the RV, heart failure and sudden death.
The reason that Havakuk and Viskin42 give to justify that the literature unanimously adopted the eponym Brugada is: "the Brugada brothers were the only authors of the original report, combined with the interesting phonetics of Brugada and the long title of this newly described entity, probably contributed to its quick adoption". This argument suggests that Havakuk and Viskin did not carefully interpret the six ECGs published by Martini et al., although in the legend of figure 2-1 B of the aforementioned manuscript is written: (B) "Presentation of 6 patients with idiopathic ventricular fibrillation, including 1 with [type-I Brugada-like pattern]". We agree that some BrS patients have been suspected of being concealed forms of ACM43. In 2019 Naoya Kataoka et al. aimed to clarify the ECG and clinical differences between BrS and ACM in long-term follow-up (mean 11.9 ± 6.3 years). A total of 50 BrS and 65 ACM patients with fatal ventricular tachyarrhythmia (VTA) were evaluated according to the revised Task Force Criteria for ACM. Based on the current diagnostic criteria concerning ECG, repolarization abnormality was positive in 2.0% and 2.6% of BrS patients at baseline and follow-up, and depolarization abnormality was positive in 6.0% and 12.8% of BrS patients at baseline and follow-up, respectively. At baseline, none of the BrS patients were definitively diagnosed with ACM. Kaplan-Meier analysis revealed that age at first VTA attack showed the same tendency between the groups. Incidence of sustained monomorphic ventricular tachycardia was significantly higher in ACM than in BrS whereas the opposite was true for VF.
Recent observations in the BrS
o Although in several BrS patients the heart is apparently structurally normal, subtle structural abnormalities mainilly in the right ventricular outflow tract (RVOT) have increasingly been reported.
o The ST segment elevation in the right pecordial leads is not persistent, frequently it is dynamic and often concealed.
o Complete RBBB is not an hallmark of BrS. Established ECG criteria of CRBBB are present only in ~16% of cases44.
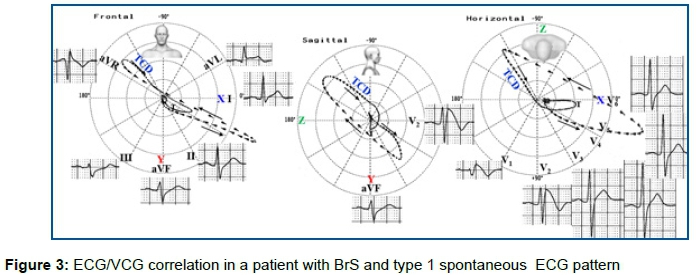
o Terminal conduction delay (TCD) of the QRS loop located near of the RVOT territory is recorded in 100% of cases and is manifested on the ECG by a significant final R wave in aVR and wide S wave in I and II. The presence of a wide and/or large S-wave in lead I is a powerful predictor of life-threatening ventricular arrhythmias in patients with BrS and no history of cardiac arrest at presentation45. In vectorcardiography (VCG), this TCD of the QRS-loop in the frontal plane(FP) is located in the upper right quadrant (near aVR) and in the right posterior quadrant in the horizontal plane(HP). This TCD of the QRS loop is manifested by terminal dashes closer to each other (≥15 dashes)46. BrS is associated with epicardial surface and interstitial fibrosis and reduced Cx43 gap junction expression in the RVOT12, which explains the TCD of the QRS loop (Figure 3).
(A) Frontal Plane(FP): prominent final R wave in aVR lead in the upper right quadrant near aVR (-150°) corresponding to the RVOT area (depolarization mechanism). TCD located on top right quadrant (terminal QRS loop comets closer to each other (≥15 dashes);
(B) Right Sagittal Plane (RSP): TCD in the upper quadrants and type 1 Brugada pattern in V2;
(C) Horizontal Plane (HP) TCD in the posterior right quadrant. Type 1 ECG Brugada ECG pattern in V1-V2.
CONCLUSIONS
We have described several nomenclatures that are closely linked to the BrS. We also put forward ECG evidence to show why a small patient series presented by Martini et al three years before the classical description of the BrS by the Brugada brothers, do not represent this entity. The type 1 Brugada ECG pattern, which is mandatory for the diagnosis of this syndrome, was absent in all but one patient in the manuscript by Martini et al.
Conflicts of interest
None declared.
Acknowledgments
We appreciated the suggestions and criticisms from Professor Arthur AM Wilde that contributed to the improvement of our text. The financial viability of the article is due to the Acre - Health Project in the Western Amazon (multiinstitutional agreement process no. 007/2015 SESACRE-UFAC-FMABC).
Contact for author´s:
Joseane Elza Tonussi Mendes: tonussijoseanecoracao@gmail.com
Kjell Nikus: kjell.nikus@sydansairaala.fi
Raimundo Barbosa-Barros: raimundobb@uol.com.br
Andrés Ricardo Pérez-Riera: riera@uol.com.br
REFERENCES
1.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391-6. [ Links ]
2.Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93(2):372-9. [ Links ]
3.Kobayashi T, Shintani U, Yamamoto T, Shida S, Isshiki N, Tanaka T, et al. Familial occurrence of electrocardiographic abnormalities of the Brugada-type. Intern Med. 1996;35(8):637-40. [ Links ]
4.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293-6. [ Links ]
5.Chokevivat V, Warintrawat S, Choprapawan C. Epidemiology of Lai Tai in Thailand. Desire Press. 1993:38-50. [ Links ]
6.Baron RC, Thacker SB, Gorelkin L, Vernon AA, Taylor WR, Choi K. Sudden death among Southeast Asian refugees. An unexplained nocturnal phenomenon. JAMA. 1983;250(21):2947-51. [ Links ]
7.Leads from the MMWR. Update: sudden unexplained death syndrome among Southeast Asian refugees--United States. JAMA. 1988;260(14):2033. [ Links ]
8.Centers for Disease C. Sudden, unexpected, nocturnal deaths among Southeast Asian refugees. MMWR Morb Mortal Wkly Rep. 1981;30(47):581-4, 9. [ Links ]
9.Parrish RG, Tucker M, Ing R, Encarnacion C, Eberhardt M. Sudden unexplained death syndrome in Southeast Asian refugees: a review of CDC surveillance. MMWR CDC Surveill Summ. 1987;36(1):43SS-53SS. [ Links ]
10.Goh KT, Chao TC, Chew CH. Sudden nocturnal deaths among Thai construction workers in Singapore. Lancet. 1990;335(8698):1154. [ Links ]
11.Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96(8):2595-600. [ Links ]
12.Nademanee K, Raju H, de Noronha SV, Papadakis M, Robinson L, Rothery S, et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J Am Coll Cardiol. 2015;66(18):1976-86. [ Links ]
13.Gaw AC, Lee B, Gervacio-Domingo G, Antzelevitch C, Divinagracia R, Jocano F, Jr. Unraveling the Enigma of Bangungut: Is Sudden Unexplained Nocturnal Death Syndrome (SUNDS) in the Philippines a Disease Allelic to the Brugada Syndrome? Philipp J Intern Med. 2011;49(3):165-76. [ Links ]
14.Vatta M, Dumaine R, Varghese G, Richard TA, Shimizu W, Aihara N, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11(3):337-45. [ Links ]
15.Guazon MP. Algunas notas sobre bangungut [in Spanish]. Revista Filipina de Medicina Y Farmacia. 1917;8:437-42. [ Links ]
16.Okada R, Kawai S. Histopathology of the conduction system in sudden cardiac death. Jpn Circ J. 1983;47(5):573-80. [ Links ]
17.Takeichi S, Nakajima K, Nakajima Y, Fujita MQ. Pathological characteristics of Pokkuri Death Syndrome; narrow circumferences of the coronary arteries in Pokkuri Death Syndrome cases. Atherosclerosis. 2008;200(1):80-2. [ Links ]
18.Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65(2):384-98. [ Links ]
19.Corrado D, Nava A, Buja G, Martini B, Fasoli G, Oselladore L, et al. Familial cardiomyopathy underlies syndrome of right bundle branch block, ST segment elevation and sudden death. J Am Coll Cardiol. 1996;27(2):443-8. [ Links ]
20.Ohe T. Idiopathic ventricular fibrillation of the Brugada type: an atypical form of arrhythmogenic right ventricular cardiomyopathy? Intern Med. 1996;35(8):595. [ Links ]
21.Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJ, Verkerk AO, de Groot JR, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation. 2005;112(18):2769-77. [ Links ]
22.Frustaci A, Russo MA, Chimenti C. Structural myocardial abnormalities in asymptomatic family members with Brugada syndrome and SCN5A gene mutation. Eur Heart J. 2009;30(14):1763. [ Links ]
23.Dhamapurkar SK, Wilson BA, Rose A, Florschutz G. Brugada syndrome and the story of Dave. Neuropsychol Rehabil. 2018;28(2):259-67. [ Links ]
24.Huang L, Tang S, Chen Y, Zhang L, Yin K, Wu Y, et al. Molecular pathological study on LRRC10 in sudden unexplained nocturnal death syndrome in the Chinese Han population. Int J Legal Med. 2017;131(3):621-8. [ Links ]
25.Ikeda T, Abe A, Yusu S, Nakamura K, Ishiguro H, Mera H, et al. The full stomach test as a novel diagnostic technique for identifying patients at risk of Brugada syndrome. J Cardiovasc Electrophysiol. 2006;17(6):602-7. [ Links ]
26.Zhao Q, Chen Y, Peng L, Gao R, Liu N, Jiang P, et al. Identification of rare variants of DSP gene in sudden unexplained nocturnal death syndrome in the southern Chinese Han population. Int J Legal Med. 2016;130(2):317-22. [ Links ]
27.Zheng J, Zheng D, Su T, Cheng J. Sudden Unexplained Nocturnal Death Syndrome: The Hundred Years' Enigma. J Am Heart Assoc. 2018;7(5). [ Links ]
28.Shimizu A, Zankov DP, Sato A, Komeno M, Toyoda F, Yamazaki S, et al. Identification of transmembrane protein 168 mutation in familial Brugada syndrome. FASEB J. 2020. [ Links ]
29.Le Scouarnec S, Karakachoff M, Gourraud JB, Lindenbaum P, Bonnaud S, Portero V, et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum Mol Genet. 2015;24(10):2757-63. [ Links ]
30.Saito YA, Strege PR, Tester DJ, Locke GR, 3rd, Talley NJ, Bernard CE, et al. Sodium channel mutation in irritable bowel syndrome: evidence for an ion channelopathy. Am J Physiol Gastrointest Liver Physiol. 2009;296(2):G211-8. [ Links ]
31.Perez-Riera AR, Baranchuk A, Zhang L, Barbosa-Barros R, de Abreu LC, Brugada P. Myotonic dystrophy and Brugada syndrome: A common pathophysiologic pathway? J Electrocardiol. 2017;50(4):513-7. [ Links ]
32.Noebels JL. Sodium channel gene expression and epilepsy. Novartis Found Symp. 2002;241:109-20; discussion 20-3, 226-32. [ Links ]
33.Priest BT, Garcia ML, Middleton RE, Brochu RM, Clark S, Dai G, et al. A disubstituted succinamide is a potent sodium channel blocker with efficacy in a rat pain model. Biochemistry. 2004;43(30):9866-76. [ Links ]
34.Vanmolkot KR, Babini E, de Vries B, Stam AH, Freilinger T, Terwindt GM, et al. The novel p.L1649Q mutation in the SCN1A epilepsy gene is associated with familial hemiplegic migraine: genetic and functional studies. Mutation in brief #957. Online. Hum Mutat. 2007;28(5):522. [ Links ]
35.Aiba T. Recent understanding of clinical sequencing and gene-based risk stratification in inherited primary arrhythmia syndrome. J Cardiol. 2019;73(5):335-42. [ Links ]
36.Campuzano O, Sarquella-Brugada G, Fernandez-Falgueras A, Cesar S, Coll M, Mates J, et al. Genetic interpretation and clinical translation of minor genes related to Brugada syndrome. Hum Mutat. 2019;40(6):749-64. [ Links ]
37.Gussak I, Bjerregaar PB. History of the Brugada syndrome. In: Antzelevitch C, Brugada P, Brugada J, Brugada R, editors. The Brugada Syndrome From Bench to Bedside. USA: Blackwell Futura; 2005 [ Links ]
38.Martini B, Nava A, Thiene G, Buja GF, Canciani B, Scognamiglio R, et al. Ventricular fibrillation without apparent heart disease: description of six cases. Am Heart J. 1989;118(6):1203-9. [ Links ]
39.Wilde AAM, Amin AS. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin Electrophysiol. 2018;4(5):569-79. [ Links ]
40.Corrado D, Basso C, Buja G, Nava A, Rossi L, Thiene G. Right bundle branch block, right precordial st-segment elevation, and sudden death in young people. Circulation. 2001;103(5):710-7. [ Links ]
41.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932-63. [ Links ]
42.Havakuk O, Viskin S. A Tale of 2 Diseases: The History of Long-QT Syndrome and Brugada Syndrome. J Am Coll Cardiol. 2016;67(1):100-8. [ Links ]
43.Corrado D, Basso C, Thiene G. Sudden cardiac death in young people with apparently normal heart. Cardiovasc Res. 2001;50(2):399-408. [ Links ]
44.Kataoka N, Nagase S, Kamakura T, Nakajima K, Wada M, Yamagata K, et al. Clinical Differences in Japanese Patients Between Brugada Syndrome and Arrhythmogenic Right Ventricular Cardiomyopathy With Long-Term Follow-Up. Am J Cardiol. 2019;124(5):715-22. [ Links ]
45.Calo L, Giustetto C, Martino A, Sciarra L, Cerrato N, Marziali M, et al. A New Electrocardiographic Marker of Sudden Death in Brugada Syndrome: The S-Wave in Lead I. J Am Coll Cardiol. 2016;67(12):1427-40. [ Links ]
46.Perez-Riera AR, Ferreira Filho C, de Abreu LC, Ferreira C, Yanowitz FG, Femenia F, et al. Do patients with electrocardiographic Brugada type 1 pattern have associated right bundle branch block? A comparative vectorcardiographic study. Europace. 2012;14(6):889-97. [ Links ]
 Correspondence:
Correspondence:
Andrés Ricardo Pérez-Riera
riera@uol.com.br
Received: June 2020
Revised: September 2020
Accepted: September 2020

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}