








Serviços Personalizados
Journal

artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Curriculum ScienTI
Curriculum ScienTIIndicadores
Compartilhar

 Permalink
PermalinkPsicologia: teoria e prática
versão impressa ISSN 1516-3687
Psicol. teor. prat. vol.18 no.1 São Paulo abr. 2016
DESENVOLVIMENTO HUMANO
Indicadores socioemocionais do espectro do autismo em pessoas com síndrome de Williams
Social and emotional indicators of the autism spectrum in individuals with Williams syndrome
Indicadores socioemocionales del espectro del autismo en individuos con síndrome de Williams
Maria Cristina Triguero Veloz TeixeiraI; Gisele da Silva BaraldiI; Deisy Ribas EmerichII; Mathias Levy de Wolinsk MiklosIII; Naiara Adorna da SilvaIII; Regina Luíza de Freitas MarinoIII; Chong Ae KimIV; Rachel Sayuri HonjoIV; Décio BrunoniV; Ana Alexandra Caldas OsórioV
I Universidade Presbiteriana Mackenzie – SP – Brasil
II Universidade de São Paulo – SP – Brasil
III Universidade Presbiteriana Mackenzie – SP – Brasil
IV Instituto da Criança, Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo – SP – Brasil
V Universidade Presbiteriana Mackenzie – SP – Brasil
Resumo
A síndrome de Williams (SW) é uma desordem genética causada pela deleção de múltiplos genes no cromossomo 7. Evidências clínicas alertam para indicadores de prejuízos socioemocionais compatíveis com Transtorno do Espectro Autista (TEA). O objetivo do estudo foi identificar indicadores socioemocionais e comportamentais compatíveis com autismo em pessoas com SW. A amostra foi composta por 30 indivíduos com diagnóstico de SW e 22 com TEA. Os instrumentos de coleta de dados foram Inventário de Comportamentos Autísticos (Autism Behavior Checklist – ABC); e Questionário de Avaliação de Autismo (Autism Screening Questionnaire – ASQ), respondidos pelos respectivos cuidadores. Foi conduzida uma análise discriminante (modelo Step Wise) para diferenciação dos grupos a partir dos itens dos inventários ABC e ASQ. O grupo de pessoas com SW apresentou um número expressivo de sinais de alterações socioemocionais, comunicativas e de comportamento compatíveis com Autismo que predominaram na fase dos quatro a cinco anos de idade.
Palavras-chave: síndrome de Williams; transtorno do espectro autista; rastreamento; competências socioemocionais; desenvolvimento.
Abstract
Williams Syndrome (WS) is a genetic disorder caused by the deletion of multiple genes on chromosome 7. Clinical evidence points to socio‑emotional alterations compatible with Autism Spectrum Disorder (ASD). The goal of this study was to identify socio‑emotional and behavioral signs compatible with ASD in individuals with WS. The sample consisted of 30 individuals with WS and 22 with ASD. The data collection instruments were Autism Behavior Checklist (ABC); and Autism Screening Questionnaire (ASQ) that were answered by the caregivers. We conducted a discriminant analysis (Step Wise) to differentiate the groups from items of the ABC and ASQ inventories. The WS group showed a large number of signs of socio‑emotional, communicative and behavioral alterations compatible with Autism that prevailed at the age of four to five years.
Keywords: Williams syndrome; autism spectrum disorder; screening; socio‑emotional competences; development.
Resumen
El Síndrome de Williams (SW) es una enfermedad genética causada por la delección de múltiplos genes en el cromosoma 7. Evidencias clínicas alertan para indicadores de afectaciones socio‑emocionales compatibles con Trastorno del Espectro del Autismo (TEA). El objetivo del estudio fue identificar indicadores socio‑emocionales y conductuales compatibles con Autismo en personas con SW. La muestra fue compuesta por 30 individuos con diagnóstico de SW y 22 con TEA. Los instrumentos de colecta de datos fueron Inventario de Conductas Autisticos (Autism Behavior Checklist – ABC); y Cuestionario de Conducta y Comunicación Social (Autism Screening Questionnaire – ASQ), respondidos por los respectivos cuidadores. Fue conducido un análisis discriminante (modelo Step Wise) para diferenciación de los grupos a partir de los itens de los inventarios ABC y ASQ. El grupo de personas con SW presentó un número expresivo de señales de alteraciones socio‑emocionales, comunicativas y de conducta compatibles con Autismo que predominaron en la fase de los cuatro a cinco años de edad.
Palabras clave: síndrome de Williams; trastorno del espectro del autismo; despistaje; competencias socio‑emocionales; desarrollo.
A síndrome de Williams (SW) é uma desordem genética causada pela microdeleção de aproximadamente 25 a 28 genes no cromossomo 7, na região 7q11.23. Alguns dos genes comprometidos na deleção são CLIP2, ELN, GTF2I, GTF2IRD1 e LIMK1, dentre outros. A incidência estimada é de 1:20.000 nascidos vivos, com prevalência estimada de 1:7.500 (Nikitina, Medvedeva, Zakharov, & Savvateeva-Popova, 2014). Apresenta um conjunto de características clínicas que geralmente auxiliam a equipe de saúde na sua identificação, a saber, dismorfismos faciais, alterações cardiovasculares (mais comumente a estenose aórtica supravalvar e outras arteriopatias), anormalidades no tecido conjuntivo, hipercalcemia infantil, deficiência no crescimento, dentre outras (Honjo, 2012). É possível a realização de um exame genético para detecção da microdeleção na região 7q11.23. Os exames convencionais mais utilizados são o método de Fluorescence in Situ Hybridization – FISH, e o método de Multiplex Ligation-dependent Probe Amplification – MLPA® (Honjo, 2012).
No fenótipo cognitivo-comportamental são descritos déficits em habilidades cognitivas, problemas emocionais e comportamentais e presença de algumas condições psiquiátricas. Especificamente nos indicadores cognitivos e de neurodesenvolvimento são descritos deficiência intelectual em níveis variados, atrasos de desenvolvimento, déficits em habilidades visoespaciais, em funções executivas como controle inibitório e memória de trabalho, bem como déficits de atenção e concentração (Nunes et al., 2013). No fenótipo de linguagem e comunicação são descritas habilidades relativamente preservadas em tarefas que avaliam habilidades de memória auditiva e verbal. No entanto, os estudos mostram também diversas alterações sintático-pragmáticas, estruturais e funcionais nas habilidades de comunicação que variam de acordo com o nível de deficiência intelectual, dificuldades para segmentar palavras, disfluências na fala e preservação de algumas habilidades expressivas de linguagem (Teixeira, Monteiro, Velloso, Kim, & Carreiro, 2010; Garayzábal-Heinze, Osório, Villaverde, & Sampaio, 2014).
No fenótipo comportamental de pessoas com SW podem ser identificados indicadores de hiperatividade e impulsividade, estereotipias corporais, gestuais e verbais, autoagressividade, excessiva responsividade social e pouca seletividade nos contatos interpessoais (inclusive com pessoas estranhas ao convívio familiar e/ou social rotineiro) e dificuldades em habilidades da vida diária básica, dentre outros (Teixeira, Carreiro, Mesquita, Khoury, & Araújo, 2012; Järvinen, Korenberg, & Bellugi, 2013; Egger et al., 2013; Waite et al., 2014).
Alguns dos contrastes e assimetrias verificados entre habilidades intelectuais e habilidades de linguagem e comunicação que são característicos da síndrome foram cunhados por Mervis como um perfil de picos e vales (peak and valley profile). Essa terminologia refere-se ao fato de, embora, por um lado, serem prejuízos cognitivos, por outro, há habilidades preservadas, e mesmo exacerbadas, como é o caso da linguagem expressiva e de algumas características dos padrões de interação e comunicação social (Mervis & John, 2010). Condições psiquiátricas como transtorno generalizado de ansiedade, fobias específicas, transtorno do déficit de atenção e hiperatividade e transtornos do espectro autista (TEA) também são verificadas em crianças, adolescentes e adultos com a síndrome (Leyfer, Woodruff-Borden, Klein-Tasman, Fricke, & Mervis, 2006). Comparadas com crianças com outros transtornos do desenvolvimento, crianças com SW são menos reservadas em relação a pessoas estranhas, costumam ser excessivas nos contatos sociais e nas habilidades expressivas de linguagem e, ao mesmo tempo que são extremamente afetuosas e amistosas, apresentam um comportamento gregário. Algumas das características de exacerbado comportamento pró-social e aumentada responsividade social provavelmente não gerariam em uma equipe de saúde o levantamento de suspeita de comorbidade com TEA em pessoas com SW. Entretanto, há evidências tanto da parte clínica como em marcadores relacionados ao endofenótipo e aspectos genéticos que alertam para a necessidade de avaliação de indicadores que possam sinalizar uma comorbidade com TEA ou prejuízos em competências socioemocionais típicas do espectro do autismo em indivíduos com SW. Isso mostra como um fenótipo caracterizado pela hipersociabilidade pode coexistir com prejuízos nas áreas de interação e comunicação social, dificuldades para fazer e manter amizades, déficits para compreender sinais relevantes no contexto de conversas sociais, habilidades sociais pobres, padrões anormais de interesses etc. (Mervis et al., 2015; Osório et. al, 2015).
O TEA é um transtorno caracterizado por sinais e sintomas relacionados com: 1. déficits persistentes na interação e na comunicação social em múltiplos contextos, por exemplo, déficits na reciprocidade socioemocional, déficits nos comportamentos comunicativos não verbais usados para interações sociais, déficits para desenvolver, manter e compreender relacionamentos; 2. presença de padrões restritos e repetitivos de comportamentos, interesses ou atividades com prejuízos no funcionamento adaptativo. Por exemplo, movimentos motores, uso de ou fala estereotipada ou repetitiva, insistência nas mesmas coisas, adesão inflexível a rotinas, respostas ritualísticas, interesses restritos e fixos, hiper ou hiporreatividade a estímulos sensoriais; 3. os sintomas devem estar presentes precocemente no período do desenvolvimento, possibilidade de verificação antes dos dois anos de vida, mas podem não se tornar plenamente manifestos quando as demandas sociais excedem as capacidades limitadas ou podem ser mascarados por estratégias aprendidas mais tarde na vida; 4. os sintomas causam prejuízo clinicamente significativo no funcionamento social, profissional e outras áreas importantes da vida da pessoa; 5. essas perturbações não são mais bem explicadas por DI ou por atraso global do desenvolvimento (American Psychiatric Association, 2014).
As principais evidências clínicas de manifestações compatíveis com TEA na SW têm sido os sinais de déficits sociocomunicativos, habilidades de cognição social (exemplo: atenção compartilhada, comportamentos de solicitação e teoria da mente), padrões comportamentais restritos e repetitivos, estereotipais e problemas de comportamento agressivo e autoagressivo (Karmiloff-Smith et al., 2012; Janes, Riby, & Rodgers, 2014; Osório et al., 2015). Até a atualidade, a maior parte dos estudos que avaliam manifestações do espectro do autismo em pessoas com SW tem focado diferentes marcadores neurais ou a utilização de instrumentos de referência para diagnóstico de TEA. Na área genética, estudo de Hibridação Genômica Comparativa por arranjos (array-CGH) detectou uma deleção na região distal do intervalo típico da SW, determinando HEMIzigose dos genes GTF2I e GTF2IRD1, confirmado por FISH. Os autores concluíram que a HEMIzigosidade de fatores de transcrição da família GTF2 poderia ser suficiente para produzir muitas das características cognitivo-comportamentais da SW, particularmente o déficit de construção visoespacial. Além disso, os autores concluíram que sintomas do autismo no caso poderiam ocorrer por causa da supressão de genes adicionais fora do intervalo típico da SW ou efeitos remotos sobre a expressão gênica em outros loci (Edelmann et al., 2007).
Na tarefa de identificar indicadores de autismo em pessoas com SW, aliados importantes seriam os instrumentos de rastreamento para autismo, ferramentas que geralmente auxiliam no reconhecimento de possíveis casos de TEA suspeitos para posteriores avaliações mais detalhadas ou para o planejamento de intervenções (Brentani et al., 2013). No Brasil existem instrumentos de rastreamento de sinais de TEA com propriedades psicométricas adequadas para esses fins. Sua ampla utilização se dá em razão de baixo custo e fácil correção, além de permitir a articulação de informações provindas de diversas fontes, tais como pais e professores (Klaiman, Fernandez-Carriba, & Hall, 2015; Nunes et al., 2011). Entretanto, estudos voltados à verificação de sinais de autismo em pessoas com SW continuam sendo escassos. Os objetivos do estudo foram identificar sinais socioemocionais, comportamentais e de socialização compatíveis com autismo em um grupo de pessoas com SW e verificar quais os itens de inventários de rastreamento de autismo que melhor identificam indicadores de TEA em pessoas com SW.
Método
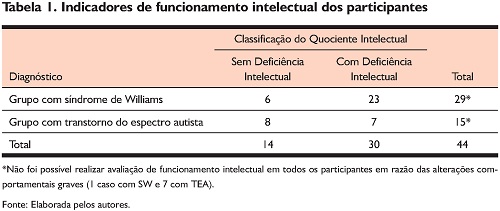
A amostra do estudo foi composta por 52 participantes distribuídos em dois grupos: a) um grupo formado por 30 crianças, adolescentes e adultos com diagnóstico genético de SW entre 8 e 41 anos (M = 17,6; DP = 7,16); e b) um grupo de 22 crianças e adolescentes com TEA entre 3 e 11 anos (M = 6,45; DP = 1,99). O diagnóstico de TEA foi estabelecido por equipe multidisciplinar de acordo com os critérios clínicos da quarta versão do Manual Estatístico e Estatístico de Transtornos Mentais (DSM-IV-TR- American Psychiatric Association, 2002). Adotou-se o DSM-IV, pois a coleta dos dados do grupo com TEA iniciou em 2013, antes da publicação oficial do DSM-V. O projeto foi aprovado pelo Comitê de Ética em Pesquisa com Seres Humanos da Universidade Presbiteriana Mackenzie, sob o protocolo n. 25707514.0.0000.0084. Respondeu aos instrumentos na condição de informantes o cuidador principal, sob o critério de exercer o cuidado, no mínimo seis horas diárias.
Para esta avaliação do funcionamento intelectual foi utilizado um dos seguintes instrumentos, dependendo da idade e do repertório verbal do participante: a Escala de Inteligência Wechsler para Crianças – WISC-III (Wechsler, 2002) – subtestes Vocabulário e Cubos para calcular o Quociente de Inteligência estimado (QI estimado), aplicado em participantes com idade entre 6 e 16 anos; a Escala de Inteligência Wechsler para Adultos – WAIS III (Wechsler, 1997) – subtestes Vocabulário e Cubos para calcular o Quociente de Inteligência estimado (QI estimado), aplicado em participantes com idade superior a 16 anos; e Teste Não Verbal de Inteligência SON-R2 ½-7 (Laros, Tellegen, Jesus, & Karino, 2015), para participantes abaixo de sete anos ou não verbais.
Instrumentos
1. Inventário de Comportamentos Autísticos (Autism Behavior Checklist - ABC) (Marteleto & Pedromônico, 2005): visa rastrear sinais e sintomas de TEA, composto por 57 itens comportamentais compatíveis com TEA distribuídos em cinco áreas diferentes: imagem corporal, sensorial, linguagem, relacional, autocuidado e interação social. Os itens são respondidos em escala dicotômica de presença ou ausência. São geradas duas pontuações: uma parcial de cada área e outra global obtida pela soma de todas as áreas do inventário. Os pontos de corte da escala estabelecem que pontuação global menor que 47 pontos é o resultado esperado para uma criança de desenvolvimento típico; 47 a 53 pontos indicam leve probabilidade para TEA; 54 a 67 pontos indicam probabilidade moderada para TEA e pontuação acima de 68 pontos indicam alterações compatíveis com TEA.
2. Questionário de Avaliação de Autismo (Autism Screening Questionnaire - ASQ) (Sato et al., 2009): visa rastrear sinais e sintomas de TEA, composto por 40 questões que avaliam interação social, linguagem e comportamentos típicos de TEA. Os itens também são respondidos em escala dicotômica de presença ou ausência. A escala de pontuação do ASQ prevê três classificações de acordo com o escore obtido: 0 a 15 pontos: normal; 16 a 21 pontos: Transtorno Invasivo do Desenvolvimento; e > de 21 pontos: autismo.
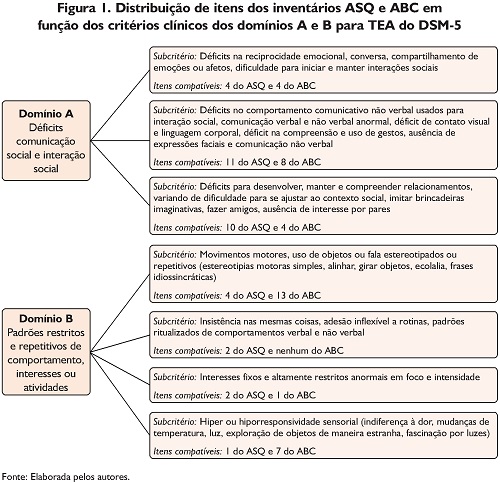
Os instrumentos ABC e ASQ não foram utilizados nas suas escalas originais de pontuação visando o rastreamento de casos suspeitos para TEA. No seu lugar, de acordo com os objetivos do estudo, foram identificados de cada um dos questionários os itens que convergiam com os critérios clínicos de TEA estabelecidos pelo DSM-V (American Psychiatric Association, 2014). Na Figura 1, apresenta-se a distribuição de itens dos inventários nos critérios clínicos em função desses domínios A e B para TEA do DSM-5 (American Psychiatric Association, 2014).
Procedimentos de coleta e análise de dados
Os dados do grupo com SW foram coletados individualmente em sala privativa de atendimento nas dependências do ambulatório de Genética Médica do Instituto da Criança do Hospital das Clínicas da Universidade de São Paulo. Os dados do grupo com TEA foram coletados individualmente em sala privativa da Secretaria de Educação da cidade de Barueri. Os dados foram organizados em um banco com o auxílio do programa SPSS (Statistical Package for the Social Sciences), versão 19.0. Foram conduzidas análises descritivas mediante uso de tabelas de frequências simples e comparação de distribuição de indicadores fenotípicos compatíveis com TEA. Foi conduzida uma análise discriminante para identificação das variáveis (itens dos inventários ABC e ASQ) com poder real de discriminação e que melhor diferenciam ou discriminam os dois grupos de participantes. O modelo utilizado para a seleção das variáveis discriminantes foi o Step Wise e os critérios adotados para a seleção de variáveis foi a estatística Wilks Lambda (λ) que expressa a relação entre a variância intragrupos e a variância total. Foi testada a matriz de variância-covariância dos grupos obtendo-se um valor de p < 0,001, o que nos permitiu concluir que existe diferença significante da matriz de covariância entre os grupos mostrando que os grupos são heterogêneos e diferentes entre eles.
Resultados e discussão
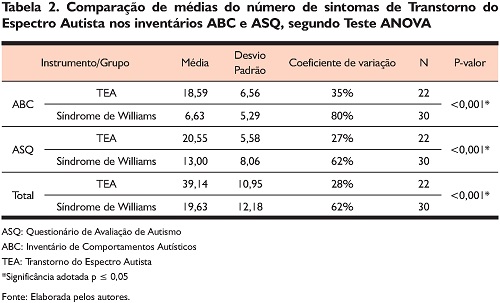
Os resultados do teste ANOVA convergiram com o esperado indicando diferenças estatisticamente significativas entre os grupos, e as médias foram superiores para o grupo com TEA. Um dado relevante da tabela são os resultados dos coeficientes de variação (CV) que foram diferentes para os grupos, sendo acima de 50% para o grupo SW, e abaixo de 50% para o grupo TEA. Do ponto de vista estatístico, é recomendável que o CV seja o mais baixo possível. A despeito da limitação estatística, há de ser observado o dado clínico que contribuiu para aumentar expressivamente o CV: de modo geral, os cuidadores reportaram que as pessoas com SW apresentaram num amplo espectro de variabilidade diversas alterações variadas em competências sociocomunicativas e de padrões de comportamento compatíveis com TEA.
Mediante o teste de Igualdade de Duas Proporções, foi realizada uma comparação da distribuição dos itens para o Domínio A (Área da comunicação e interação social entre os grupos). Não foram verificadas diferenças estatisticamente significativas entre os grupos em um número expressivo de itens, sendo esse um dado clínico importante. Em ordem decrescente, os percentuais relativos ao grupo com SW em que se verificaram os maiores indicadores de autismo e sem diferença estatisticamente significativa com o grupo com TEA, todos referentes ao instrumento ASQ, foram: quando estava com 4 ou 5 anos não brincava cooperativamente em jogos de grupo, como esconde-esconde e jogos de bola (80%); quando estava com 4 ou 5 anos, não costumava brincar de faz de conta com outra criança de forma que o cuidador percebia que eles estavam entendendo ser uma brincadeira (76,7%); quando estava com 4 ou 5 anos, apresentava dificuldades para participar espontaneamente e/ou tentar imitar ações em jogos sociais, tais como Polícia e Ladrão ou Pega-Pega (70%); não tem um amigo em particular ou melhor amigo (50%); quando tinha 4 ou 5 anos, não apontava as coisas ao redor espontaneamente apenas para mostrar e não porque as desejava (50%); quando tinha 4 ou 5 anos, não costumava dividir coisas com as pessoas, além de alimentos (46,7%); quando tinha 4 ou 5 anos, não parecia interessado em outras crianças da mesma idade que ele não conhecia (40%); quando tinha 4 ou 5 anos, não costumava querer que o cuidador participasse de algo que o estava divertindo (33,3%); diz frases estranhas inventadas por ele ou copia frases de outras pessoas e as repete da mesma maneira (33,3%); até onde o cuidador percebe, a expressão facial dele(a) não parece apropriada à situação particular (23,3%); uso de perguntas inapropriadas ou declarações em contextos sociais inadequados (23,3%); incapacidade para manter diálogos ou alternância durante conversas (16,7%).
Associa-se à SW um fenótipo de hipersociabilidade oposto ao fenótipo de evitação social característico do TEA (Järvinen et al., 2013). No entanto, merecem cuidados especiais diversos indicadores qualitativos clínicos encontrados que sugerem que os participantes com SW apresentaram e/ou apresentam atualmente em níveis variados dificuldades na interação e comunicação social, dificuldades para fazer e manter amizades e déficits para compreender sinais relevantes do contexto durante conversas sociais, dentre outros domínios tipicamente afetados em pessoas com TEA (Van der Fluit, Gaffrey, & Klein-Tasman, 2012).
De acordo com as respostas dos cuidadores aos inventários, verificamos que ao longo dos primeiros quatro ou cinco anos de idade o grupo de participantes com SW apresentou indicadores de afetação em várias competências socioemocionais como incapacidade de imitação, de partilha emocional e de atenção compartilhada (avaliados predominantemente no inventário ASQ). São dados clínicos indicativos de que, nesses primeiros cinco anos, os indivíduos com SW não atingiam diversos marcos esperados de desenvolvimento em relação a habilidades sociais e sensibilidade a determinadas pistas do contexto social, assim como ocorre em pessoas com TEA (Mervis et al., 2015).
Apesar desses prejuízos socioemocionais descritos no parágrafo anterior, um dado relevante relatado pelo cuidador informante é que nessas pessoas com SW da amostra, ao que parece, há também outras habilidades preservadas; por exemplo, discriminar informações sociais do contexto como rostos, gestos e vozes de pessoas. Um dos itens do inventário ASQ que avalia alguns desses indicadores de comunicação, percepção de emoções e motivação social é o item 10 (Até onde você percebe, a expressão facial dele(a) geralmente parece apropriada à situação particular). No item, 100% dos participantes do grupo com SW não apresentam problemas se comparado a 55% dos indivíduos com TEA nos que se informa a presença de problema. Entretanto, o dado deve ser interpretado com cautela, em razão da complexidade de fatores neurobiológicos imbricados, sendo alguns deles associados a manifestações do endofenótipo. Estudos focados na avaliação de habilidades de reconhecimento de faces na SW mostram resultados diferentes de acordo com a faixa etária e especificidades das tarefas de reconhecimento (Isaac & Lincoln, 2011). Dados recentes refutaram estudos previamente publicados que focavam o reconhecimento global de faces e nos quais se concluía que o processamento de faces na SW estava intacto, e demonstraram que crianças com SW desde idades precoces (idade média de 26 meses) já apresentam falhas nas estratégias de processamento de faces, quando comparadas com o grupo controle pareados por idade (DSouza et al., 2015). Os autores reportaram a ocorrência de processamentos atípicos, tanto em tarefas de reconhecimento de características isoladas de faces como em tarefas de reconhecimento de faces em situações reconfiguradas (DSouza et al., 2015). Provavelmente, esse seja um dos fatores que expliquem a responsividade social e a falta de discriminação de estímulos em contexto social de interação com pobres habilidades sociais (Järvinen et al., 2013).
A responsividade social presente na SW (Järvinen & Bellugi, 2014) é instigante do ponto de vista clínico-genético. A SW é uma desordem genética cuja etiologia está bem estabelecida, diferente de TEA (Nikitina et al., 2014). Daí a ênfase que atualmente é dada ao estudo de indicadores do denominado fenótipo social da SW em contraste com fenótipo não social dos TEA (Crespi & Hurd, 2014). A duplicação da região 7q11.23, ou seja, da mesma região deletada na SW, apresenta um fenótipo com características de TEA, dentre outras alterações comportamentais (Mervis et al., 2015).
Uma vez conduzidas as análises para o Domínio A, realizou-se o Teste de Igualdade de Duas Proporções, para comparar os grupos a respeito da distribuição de itens compatíveis com o Domínio B (Padrões restritos e repetitivos de comportamento). Em ordem decrescente, os percentuais relativos ao grupo com SW em que se verificaram os maiores indicadores de autismo e sem diferença estatisticamente significativa com o grupo com TEA, todos referentes ao instrumento ASQ, foram: costuma se interessar mais por partes de um objeto ou brinquedo, mais do que usá-lo na sua função original (40%); interesses específicos, apropriados para sua idade e para seu grupo de colegas, porém, estranhos pela intensidade do interesse (33,3%); ações muito particulares da pessoa, em determinada ordem ou seguindo rituais que obriga a outras pessoas a fazer (30%); tem um objeto (que não é um brinquedo macio ou cobertor) que carrega por toda parte (30%); interesses especiais que parecem esquisitos a outras pessoas, por exemplo, semáforos (23,3%); costuma, de maneira estranha, olhar, sentir, examinar, escutar, provar ou cheirar objetos ou pessoas (23,3%). Estudo brasileiro de Nunes e colaboradores também verificou sinais de alteração comportamental típica de Autismo em 42% de 31 pacientes com SW (Nunes et al., 2013). Entretanto, deve ser levantada outra hipótese em relação a que essas alterações comportamentais podem ser também decorrentes da condição de rebaixamento intelectual de 80% do grupo com SW, como mostrado em estudos anteriores, e não apenas a TEA (Hahn, Fidler & Hepburn, 2014).
Na Tabela 3 constam os resultados da Análise Discriminante para identificação das variáveis (sinais de TEA de acordo com itens dos inventários ABC e ASQ) com poder real de discriminação entre os dois grupos de participantes. De acordo com o modelo Step Wise e a estatística Wilks Lambda (λ) adotada, na matriz de variância-covariância dos grupos, foi obtido um valor de p < 0,001. De acordo com esse valor, conclui-se que existe diferença significativa da matriz de covariância entre os grupos mostrando que eles são heterogêneos e diferentes entre si em função de 11 variáveis (sinais de TEA de acordo com itens dos inventários ABC e ASQ). Os valores de Wilks lambda, próximos de zero, indicaram que nesses sinais a maioria da variabilidade total pode ser atribuída a diferenças entre as médias dos grupos, havendo uma pequena variabilidade intragrupos se comparada à variabilidade total.
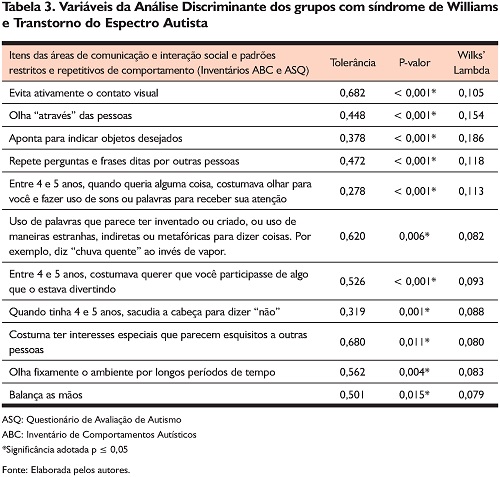
Para testar a significância da função discriminante, foram verificados o Teste de Wilks Lambda e o Valor Próprio (Eigenvalue) e a variabilidade explicada por este. Na Tabela 4, verifica-se um valor próprio superior a 1 (Eigenvalue = 13,8), é alto, e responsável por 100% da variância total que corresponde a 100% da variabilidade explicada. Também o teste de Wilks Lambda, com valor de 0,068, demonstrou que a função discriminante estatisticamente é altamente significativa (p < 0,001), mostrando que existe uma elevada relação dos 11 sinais selecionados na discriminação ou diferenciação dos grupos.
A função discriminante testada para os 11 itens do ABC e ASQ foi altamente significativa na diferenciação dos grupos a partir desses sinais (Tabela 3). Esses especificadores de TEA parecem ser os sinais que realmente diferenciaram os participantes com SW dos que apresentam TEA no contexto da amostra. No entanto, em razão de limitações decorrentes do tamanho amostral, não é possível afirmar que esses sejam os únicos e melhores itens para essa discriminação de uma pessoa com SW de outra com TEA. Em função do tipo de instrumento utilizado e das análises conduzidas no estudo, os sinais encontrados na Análise Discriminante não permitem efetuar julgamentos diagnósticos em relação a TEA no grupo com SW. A despeito de tais limitações, o presente trabalho indica áreas que devem ser cuidadosamente analisadas por profissionais de saúde para a avaliação de autismo em pessoas com SW. Observe-se que alguns dos sinais indicadores relacionam-se fortemente ao domínio da comunicação e interação social (critério diagnóstico do Domínio A da atual classificação diagnóstica de TEA do DSM-5) (American Psychiatric Association, 2014), tais como: evita ativamente o contato visual, olha através das pessoas; entre 4 e 5 anos apresentava dificuldades para expressar que o cuidador participasse de algo que o estava divertindo; entre 4 e 5 anos, quando queria alguma coisa, tinha dificuldades para olhar para o cuidador e fazer uso de sons ou palavras para receber sua atenção. Já os sinais restantes pertencem ao critério diagnóstico do Domínio B. Por exemplo, repete perguntas e frases ditas por outras pessoas; emprego de palavras que parece ter inventado ou criado ou uso de maneiras estranhas, indiretas ou metafóricas para dizer coisas; costuma ter interesses especiais que parecem esquisitos a outras pessoas; olha fixamente o ambiente por longos períodos de tempo e balança as mãos.
Os altos percentuais de sinais relatados pelos cuidadores sobre comportamentos ocorridos quando o participante estava com idade entre os 4 e 5 anos são sugestivos de que, provavelmente, esses teriam se beneficiado com intervenções que focassem o envolvimento interpessoal, o desenvolvimento de habilidades adequadas de imitação, o desenvolvimento de habilidades verbais e não verbais de comunicação e a estimulação de competências para jogos interativos e cooperativos, dentre outros, testadas com sucesso em intervenções junto a crianças com TEA (Rogers & Dawson, 2010).
Conclusão
No grupo com SW foi verificado um número expressivo de sinais de alterações socioemocionais, comunicativas e de comportamento compatíveis com autismo. Os instrumentos de coleta de dados permitem avaliar sinais e sintomas presentes na atualidade, assim como em períodos precoces do desenvolvimento, por exemplo entre 4 e 5 anos. De fato, de acordo com o relato dos informantes, um número expressivo de sinais predominou na fase dos 4 a 5 anos de idade, como problemas na compreensão e uso de gestos, dificuldades para iniciar e manter interações sociais, déficits para imitar brincadeiras, dificuldades para fazer amigos, déficits de interesse social para se relacionar com outras crianças da mesma idade desconhecidas ou estranhas. Trata-se de um conjunto de sinais que, pelas especificidades dos instrumentos utilizados, não nos permite afirmar se esses indicadores estão presentes nos participantes avaliados na atualidade, por ocasião do estudo. É uma limitação do trabalho e, sendo assim, estudos futuros poderiam adotar desenhos longitudinais para acompanhamento desses sinais e sintomas. Com isso, seria possível verificar a evolução desses ao longo do desenvolvimento, inclusive na idade adulta. De outro lado, a SW é considerada uma síndrome rara cujo diagnóstico é complexo. Contudo, nos últimos dez anos crianças no Brasil têm recebido cada vez mais diagnósticos precoces. Por isso, espera-se que nas próximas décadas possam ser conduzidos esses estudos que permitirão traçar um perfil comportamental e socioemocional ao longo do desenvolvimento. Outros sinais mais típicos de autismo e, geralmente, observados em pessoas com TEA com maior comprometimento socioadaptativo, não foram relatados pelos cuidadores, como evitar o contato visual, não apontar para indicar objetos desejados, invenção de frases estranhas, balançar as mãos, dentre outros.
A técnica de Análise Discriminante utilizada no estudo permitiu identificar, a partir dos itens dos instrumentos ABC e ASQ, um conjunto de onze sinais com poder real de discriminação entre o grupo com SW e o grupo com TEA (seis itens ou sinais pertencem ao instrumento ABC e cinco ao ASQ). Ressaltando como limitação que, como todo estudo que utiliza instrumentos baseados no relato do cuidador na condição de informante, os dados poderão ter o viés da percepção desse. A verificação de condições psiquiátricas como TEA em grupos sindrômicos de etiologia genética está bem estabelecida, tanto na pesquisa básica quanto na clínica. Entretanto, recomenda-se para estudos futuros testar outras funções discriminantes, por exemplo, avaliar regras para a alocação de novos indivíduos com SW na condição de suspeita de sinais ou comprometimentos do espectro do autismo. Para trabalhos futuros, também se recomenda a utilização de diferentes grupos etários nos que se esperam marcos de desenvolvimento relativamente mais homogêneos.
Os instrumentos utilizados evidenciaram que o grupo com SW apresenta diferentes indicadores de comprometimento característicos do espectro do autismo que provavelmente estão impactando negativamente na qualidade de vida individual e familiar. A avaliação de crianças com SW nos primeiros cinco anos de vida pode permitir intervenções adequadas focadas na estimulação de habilidades de iniciativa para interações e envolvimento social, visando aproveitar a plasticidade do período infantil. A partir dos resultados do estudo recomendamos que, para crianças com SW até os 6 anos de idade, sejam utilizados protocolos de avaliação clínica que permitam não só o rastreamento de sinais de TEA, como também posteriores avaliações compreensivas dos casos suspeitos com uso de instrumentos de referência para confirmar diagnóstico de autismo. Essas ações possibilitarão o planejamento, em idades precoces, de intervenções com foco desenvolvimentista e comportamental.
Referências
American Psychiatric Association. (2014). Manual diagnóstico e estatístico de transtornos mentais-DSM 5. Porto Alegre: Artmed. [ Links ]
Brentani, H., De Paula, C. S., Bordin, D., Rolim, D., Sato, F., Portolese, J., & Mccracken, J. T. (2013). Autism spectrum disorders: An overview on diagnosis and treatment. Revista Brasileira de Psiquiatria, 35, 62-72. [ Links ]
Crespi, B. J., & Hurd, P. L. (2014). Cognitive-behavioral phenotypes of Williams syndrome are associated with genetic variation in the GTF2I gene, in a healthy population. BMC Neurosci, 15, 127. [ Links ]
DSouza, D., Cole, V., Farran, K. E., Brown, H. J., Humphreys, K., Howard, J., Rodic, M., Dekker, M. T., DSouza, H., & Karmiloff-Smith, A. (2015). Face processing in Williams syndrome is already atypical in infancy. Frontiers in Psychology, 6(760), 1-9. [ Links ]
Edelmann, L., Prosnitz, A., Pardo, S., Bhatt, J., Cohen, N., Lauriat, T., & Mcinnes, L. A. (2007). An atypical deletion of the Williams-Beuren syndrome interval implicates genes associated with defective visuospatial processing and autism. Journal of Medical Genetics, 44(2), 136-143.
Egger, J. I., Wingbermühle, E., Verhoeven, W., Dijkman, M., Radke, S., De Bruijn, E. R., & Koolen, D. (2013). Hypersociability in the behavioral phenotype of 17q21. 31 microdeletion syndrome. American Journal of Medical Genetics, Part A, 161(1), 21-26. [ Links ]
Garayzábal-Heinze, E., Osório, A., Villaverde, M. L., & Sampaio, A. (2014). Concrete and relational vocabulary: Comparison between Williams and Smith-Magenis syndromes. Research in Developmental Disabilities, 35, 3365-3371. [ Links ]
Hahn, L. J., Fidler, D. J., & Hepburn, S. L. (2014). Adaptive behavior and problem behavior in young children with Williams syndrome. American Journal on Intellectual and Developmental Disabilities, 119(1), 49-63. [ Links ]
Honjo, R. S. (2012). Detecção da microdeleção 7q11.23 por MLPA e estudo clínico dos pacientes com síndrome de Williams-Beuren. Tese de doutorado, Faculdade de Medicina da Universidade de São Paulo, Programa de Pediatria, São Paulo, SP, Brasil. [ Links ]
Isaac, L., & Lincoln, A. (2011). Featural versus configural face processing in a rare genetic disorder: Williams syndrome. Journal of Intellectual Disability Research, 55(11), 1034-1042. [ Links ]
Janes, E., Riby, D. M., & Rodgers, J. (2014). Exploring the prevalence and phenomenology of repetitive behaviours and abnormal sensory processing in children with Williams Syndrome. Journal of Intellectual Disability Research, 58, 746-757. [ Links ]
Järvinen, A., & Bellugi, U. (2014). Toward a deeper characterization of the social phenotype of Williams syndrome: The association between personality and social drive. Research in Developmental Disabilities, 35(8), 1838-1849. [ Links ]
Järvinen, A., Korenberg, J. R., & Bellugi, U. (2013). The Social Phenotype of Williams Syndrome. Current Opinion Neurobiology, 23(3), 414-422. [ Links ]
Karmiloff-Smith, A., Broadbent, H., Farran, E. K., Longhi, E., Dsouza, D., Metcalfe, K., & Sansbury, F. (2012). Social cognition in Williams syndrome: genotype/phenotype insights from partial deletion patients. Frontiers in Psychology, 30(3), 168. [ Links ]
Klaiman, C., Fernandez-Carriba, S., & Hall, C. (2015). Assessment of autismo Across the Lifespan: A Way Forward. Current Developmental Disorder Report, 2, 84-98. [ Links ]
Laros, J. A., Tellegen, P. J., Jesus, G. R., de, & Karino, C. A. (2015). SON-R 21/2-7[a], Teste não-verbal de inteligência. Manual com normatização e validação brasileira. São Paulo: Editora CETEPP. [ Links ]
Leyfer, O. T., Woodruff-Borden, J., Klein-Tasman, B. P., Fricke, J. S., & Mervis, C. B. (2006). Prevalence of psychiatric disorders in 4 to 16 years-olds with Williams syndrome. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 141(6), 615-622. [ Links ]
Marteleto, M. R. F., & Pedromônico, M. R. M. (2005). Validade do inventário de comportamentos autísticos (ICA): estudo preliminar. Revista Brasileira de Psiquiatria, 27(4), 295-301. [ Links ]
Mervis, C. B., & John, A. E. (2010). Cognitive and behavioral characteristics of children with Williams syndrome: Implications for intervention approaches. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 154(2), 229-248. [ Links ]
Mervis, C. B., Klein-Tasman, B. P., Huffman, M. J., Velleman, S. L., Pitts, C. H., Henderson, D. R., Woodruff-Borden, J., Morris, C. A., & Osborne, L. R. (2015). Children with 7q11.23 duplication syndrome: Psychological characteristics. American Journal of Medical Genetics Part A, 167(7), 1436-1450. [ Links ]
Nikitina, E. A., Medvedeva, A. V., Zakharov, G. A., & Savvateeva-Popova, E. V. (2014). Williams syndrome as a model for elucidation of the pathway genes - the brain - cognitive functions: genetics and epigenetics. Acta Naturae, 6(1), 9-22. [ Links ]
Nunes, M. M., Honjo, R. S., Dutra, R. L., Amaral, V. A. S., Oh, H. K., Bertola, D. R., Albano, L. M. J., Assumpção Junior, F. B., & Kim, C. A. (2011). High frequency of autistic traits in Williams-Beuren patients. Pediatria, 33, 81-88. [ Links ]
Nunes, M. M., Honjo, R. S., Dutra, R. L., Amaral, V. S., Oh, H. K., Bertola, D. R., Albano, L. M. J., Assumpção, J. F. B., Kim, C. A., & Teixeira, M. C. T. V. (2013). Spatial abilities in children and adults with Williams syndrome. Universita Psychologica, 12(2), 581-589. [ Links ]
Osório, A., Sampaio, A., Regueiro, R. M., Heinze, E. G., Carracedo, A., & Prieto, M. F. (2015). Autism spectrum symptoms in Smith-Magenis syndrome and Williams syndrome: comparisons and contrasts. International Journal of Developmental Disabilities, 61(1), 49-55. [ Links ]
Rogers, S. J., & Dawson, G. (2010). Intervenção precoce em crianças com Autismo. Lisboa: Lidel – Edições Técnicas. [ Links ]
Sato, F. P., Paula, C. S., Lowenthal, R., Nakano, E. Y., Brunoni, D., Schwartzman, J. S., & Mercadante, M. T. (2009). Instrument to screen cases of pervasive developmental disorder: a preliminary indication of validity. Revista Brasileira de Psiquiatria, 31(1), 30-33. [ Links ]
Teixeira, M. C. T. V., Carreiro, L. R. R., Mesquita, M. L. G., Khoury, L. P., & Araújo, M. V. (2012). Mood Disorders in individuals with genetic syndromes and intellectual disability. In M. Juruena. Clinical, research and treatment approaches to affective disorders (pp. 49-72), Croatia: In Tech. [ Links ]
Teixeira, M. C. T. V., Monteiro, C. R. C., Velloso, R. D. L., Kim, C., & Carreiro, L. R. R. (2010). Behavioral and cognitive phenotype of children and adolescents with Williams-Beuren Syndrome. Pró-Fono Revista de Atualização Científica, 22(3), 215-220. [ Links ]
Van Der Fluit, F., Gaffrey, M. S., & Klein-Tasman, B. P. (2012). Social cognition in Williams syndrome: relations between performance on the social attribution task and cognitive and behavioral characteristics. Front Psychol., 3, 197. [ Links ]
Waite, J., Heald, M., Wilde, L., Woodcock, K., Welham, A., Adams, D., & Oliver, C. (2014). The importance of understanding the behavioural phenotypes of genetic syndromes associated with intellectual disability. Pediatrics and Child Health, 24(10), 1-5. [ Links ]
Wechsler, D. (1997). Wechsler adult intelligence scale. (WAIS-III). San Antonio, Tex: Harcourt Assessment. [ Links ]
Wechsler, D. (2002). Escala de Inteligência de Wechsler para Crianças. [Manual]. Lisboa: Cegoc. [ Links ]
 Endereço para correspondência:
Endereço para correspondência:
Maria Cristina Triguero Veloz Teixeira
Programa de Pós‑graduação em Distúrbios da Universidade Presbiteriana Mackenzie
Rua da Consolação, 896, Consolação
São Paulo – SP – Brasil. CEP: 01302907
E-mail: cris@teixeira.org
Submissão: 22.8.2015
Aceitação: 19.11.2015